|
OpenMS
|
Detects peptide pairs in LC-MS data and determines their relative abundance.
| pot. predecessor tools | → FeatureFinderMultiplex → | pot. successor tools |
|---|---|---|
| FileConverter | IDMapper | |
| FileFilter |
FeatureFinderMultiplex is a tool for the fully automated analysis of quantitative proteomics data. It detects pairs of isotopic envelopes with fixed m/z separation. It requires no prior sequence identification of the peptides and works on both profile or centroided spectra. In what follows we outline the algorithm.
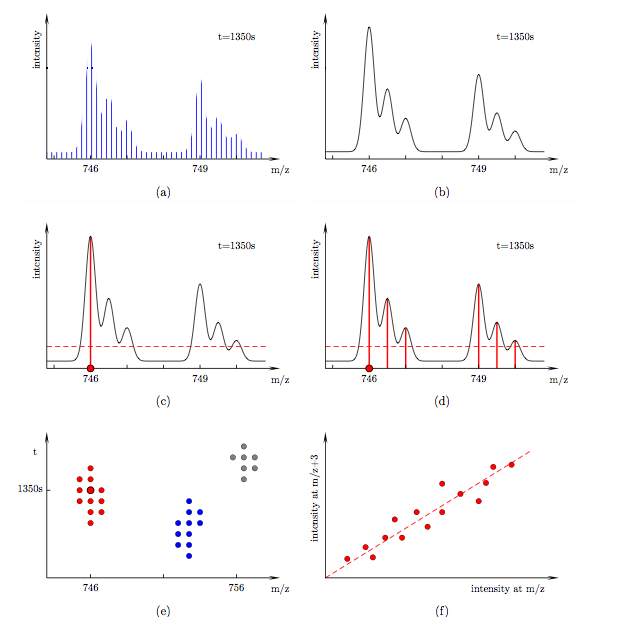
Algorithm The algorithm is divided into three parts: filtering, clustering and linear fitting, see Fig. (d), (e) and (f). In the following discussion let us consider a particular mass spectrum at retention time 1350 s, see Fig. (a). It contains a peptide of mass 1492 Da and its 6 Da heavier labelled counterpart. Both are doubly charged in this instance. Their isotopic envelopes therefore appear at 746 and 749 in the spectrum. The isotopic peaks within each envelope are separated by 0.5. The spectrum was recorded at finite intervals. In order to read accurate intensities at arbitrary m/z we spline-fit over the data, see Fig. (b). We would like to search for such peptide pairs in our LC-MS data set. As a warm-up let us consider a standard intensity cut-off filter, see Fig. (c).
Scanning through the entire m/z range (red dot) only data points with intensities above a certain threshold pass the filter. Unlike such a local filter, the filter used in our algorithm takes intensities at a range of m/z positions into account, see Fig. (d). A data point (red dot) passes if
Let us now filter not only a single spectrum but all spectra in our data set. Data points that pass the filter form clusters in the t-m/z plane, see Fig. (e). Each cluster corresponds to the mono-isotopic mass trace of the lightest peptide of a SILAC pattern. We now use hierarchical clustering methods to assign each data point to a specific cluster. The optimum number of clusters is determined by maximizing the silhouette width of the partitioning. Each data point in a cluster corresponds to three pairs of intensities (at [m/z, m/z+3], [m/z+0.5, m/z+3.5] and [m/z+1, m/z+4]). A plot of all intensity pairs in a cluster shows a clear linear correlation, see Fig. (f). Using linear regression we can determine the relative amounts of labelled and unlabelled peptides in the sample.
The command line parameters of this tool are:
FeatureFinderMultiplex -- Determination of peak ratios in LC-MS data
Full documentation: http://www.openms.de/doxygen/release/3.5.0/html/TOPP_FeatureFinderMultiplex.html
Version: 3.5.0 Dec 11 2025, 12:37:51, Revision: c1370fb
To cite OpenMS:
+ Pfeuffer, J., Bielow, C., Wein, S. et al.. OpenMS 3 enables reproducible analysis of large-scale mass spec
trometry data. Nat Methods (2024). doi:10.1038/s41592-024-02197-7.
Usage:
FeatureFinderMultiplex <options>
Options (mandatory options marked with '*'):
-in <file>* LC-MS dataset in either centroid or profile mode (valid formats:
'mzML')
-out <file> Output file containing the individual peptide features. (valid
formats: 'featureXML')
-faims_merge_features <true/false> For FAIMS data with multiple compensation voltages: Merge features
representing the same analyte detected at different CV values
into a single feature. Only features with DIFFERENT FAIMS CV value
s are merged (same CV = different analytes). Has no effect on non-
FAIMS data. (default: 'true') (valid: 'true', 'false')
algorithmic parameters:
-algorithm:labels <text> Labels used for labelling the samples. If the sample is unlabelled
(i.e. you want to detect only single peptide features) please
leave this parameter empty. [...] specifies the labels for a singl
e sample. For example
[][Lys8,Arg10] ... SILAC
[][Lys4,Arg6][Lys8,Arg10] ... triple-SILAC
[Dimethyl0][Dimethyl6] ... Dimethyl
[Dimethyl0][Dimethyl4][Dimethyl8] ... triple Dimethyl
[ICPL0][ICPL4][ICPL6][ICPL10] ... ICPL (default: '[][Lys8,A
rg10]')
-algorithm:charge <text> Range of charge states in the sample, i.e. min charge : max charge
. (default: '1:4')
-algorithm:rt_typical <value> Typical retention time [s] over which a characteristic peptide
elutes. (This is not an upper bound. Peptides that elute for longe
r will be reported.) (default: '40.0') (min: '0.0')
-algorithm:rt_band <value> The algorithm searches for characteristic isotopic peak patterns,
spectrum by spectrum. For some low-intensity peptides, an importan
t peak might be missing in one spectrum but be present in one of
the neighbouring ones. The algorithm takes a bundle of neighbourin
g spectra with width rt_band into account. For example with rt_ban
d = 0, all characteristic isotopic peaks have to be present in
one and the same spectrum. As rt_band increases, the sensitivity
of the algorithm but also the likelihood of false detections incre
ases. (default: '0.0') (min: '0.0')
-algorithm:rt_min <value> Lower bound for the retention time [s]. (Any peptides seen for a
shorter time period are not reported.) (default: '2.0') (min: '0.0
')
-algorithm:mz_tolerance <value> M/z tolerance for search of peak patterns. (default: '6.0') (min:
'0.0')
-algorithm:mz_unit <choice> Unit of the 'mz_tolerance' parameter. (default: 'ppm') (valid:
'Da', 'ppm')
-algorithm:intensity_cutoff <value> Lower bound for the intensity of isotopic peaks. (default: '1000.0
') (min: '0.0')
-algorithm:peptide_similarity <value> Two peptides in a multiplet are expected to have the same isotopic
pattern. This parameter is a lower bound on their similarity.
(default: '0.5') (min: '-1.0' max: '1.0')
-algorithm:averagine_similarity <value> The isotopic pattern of a peptide should resemble the averagine
model at this m/z position. This parameter is a lower bound on
similarity between measured isotopic pattern and the averagine
model. (default: '0.4') (min: '-1.0' max: '1.0')
-algorithm:missed_cleavages <number> Maximum number of missed cleavages due to incomplete digestion.
(Only relevant if enzymatic cutting site coincides with labelling
site. For example, Arg/Lys in the case of trypsin digestion and
SILAC labelling.) (default: '0') (min: '0')
Common TOPP options:
-ini <file> Use the given TOPP INI file
-threads <n> Sets the number of threads allowed to be used by the TOPP tool
(default: '1')
-write_ini <file> Writes the default configuration file
--help Shows options
--helphelp Shows all options (including advanced)
INI file documentation of this tool:
 1.9.1
1.9.1 
