![]()
User Tutorial
![]()

![]()
User Tutorial
![]()
The OpenMS Developers
May 9, 2021
Creative Commons Attribution 4.0 International
(CC BY 4.0)
 folder, on the USB stick that came with this tutorial, or released online on our
GitHub repository OpenMS/Tutorials.
folder, on the USB stick that came with this tutorial, or released online on our
GitHub repository OpenMS/Tutorials.
Before we get started we will install OpenMS and KNIME. If you take part in a training session you will have likely received an USB stick from us that contains the required data and software. If we provide laptops with the software you may of course skip the installation process and continue reading the next section.
Please choose the directory that matches your operating system and execute the installer.
For example for Windows you call


 ).
).on macOS you call


and follow the instructions. For the OpenMS installation on macOS, you need to accept the license drag and drop the OpenMS folder into your Applications folder.
Note: Due to increasing security measures for downloaded apps (e.g. path randomization) on
macOS you might need to open TOPPView.app and TOPPAS.app while holding
 and
accept the warning. If the app still does not open, you might need to move them from
and
accept the warning. If the app still does not open, you might need to move them from
 to
e.g. your Desktop and back.
to
e.g. your Desktop and back.
On Linux you can extract KNIME to a folder of your choice and for TOPPView you need to install OpenMS via your package manager or build it on your own with the instructions under www.openms.de/documentation.
Note: If you have installed OpenMS on Linux or macOS via your package manager (for instance by installing the OpenMS-2.6.0-Linux.deb package), then you need to set the OPENMS_DATA_PATH variable to the directory containing the shared data (normally /usr/share/OpenMS). This must be done prior to running any TOPP tool.
If you are working through this tutorial at home you can get the installers under the following links:
Choose the installers for the platform you are working on.
Each MS instrument vendor has one or more formats for storing the acquired data. Converting these data into an open format (preferably mzML) is the very first step when you want to work with open-source mass spectrometry software. A freely available conversion tool is MSConvert, which is part of a ProteoWizard installation. All files used in this tutorial have already been converted to mzML by us, so you do not need to perform the data conversion yourself. However, we provide a small raw file so you can try the important step of raw data conversion for yourself.
Note: The OpenMS installation package for Windows automatically installs ProteoWizard, so you do not need to download and install it separately. Due to restrictions from the instrument vendors, file format conversion for most formats is only possible on Windows systems. In practice, performing the conversion to mzML on the acquisition PC connected to the instrument is usually the most convenient option.
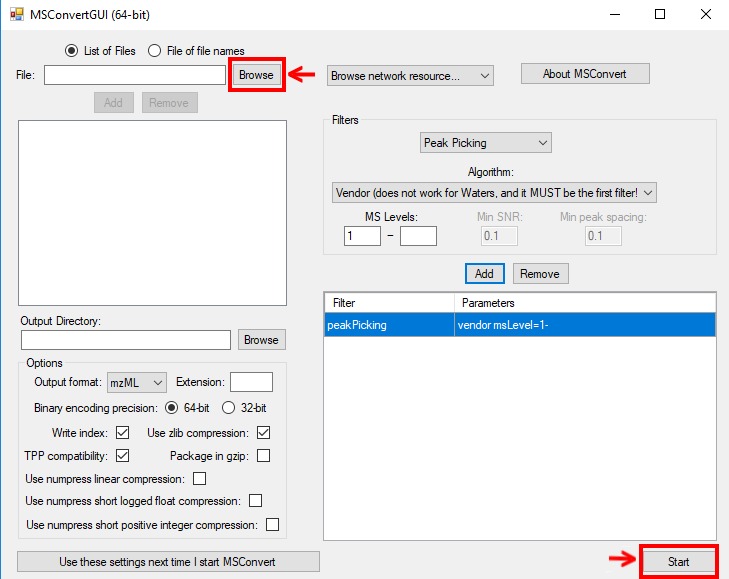
To convert raw data to mzML using ProteoWizard you can either use MSConvertGUI (a
graphical user interface) or msconvert (a simple command line tool). Both tools are available
in:  . You
can find a small RAW file on the USB stick
. You
can find a small RAW file on the USB stick  .
.
MSConvertGUI (see Fig. 1) exposes the main parameters for data conversion in a convenient graphical user interface.
The msconvert command line tool has no user interface but offers more options than the application MSConvertGUI. Additionally, since it can be used within a batch script, it allows converting large numbers of files and can be much more easily automatized.
To convert and pick the file raw_data_file.RAW you may write:

in your command line.
To convert all RAW files in a folder may write:

Note: To display all options you may type  .
Additional information is available on the ProteoWizard web page.
.
Additional information is available on the ProteoWizard web page.
Recently the open-source platform independent ThermoRawFileParser tool has been developed. While Proteowizard and MSConvert are only available for Windows systems this new tool allows to also convert raw data on Mac or Linux.
Note: To learn more about the ThermoRawFileParser and how to use it in KNIME see Section 2.4.7
Visualizing the data is the first step in quality control, an essential tool in understanding the data, and of course an essential step in pipeline development. OpenMS provides a convenient viewer for some of the data: TOPPView.
We will guide you through some of the basic features of TOPPView. Please familiarize yourself with the key controls and visualization methods. We will make use of these later throughout the tutorial. Let’s start with a first look at one of the files of our tutorial data set. Note that conceptually, there are no differences in visualizing metabolomic or proteomic data. Here, we inspect a simple proteomic measurement:
 on macOS)
on macOS)
 , navigate to the directory where you copied the contents of the USB stick to, and
select
, navigate to the directory where you copied the contents of the USB stick to, and
select  . This file contains only a reduced LC-MS map 1
of a label-free proteomic platelet measurement recorded on an Orbitrap velos. The
other two mzML files contain technical replicates of this experiment. First, we want
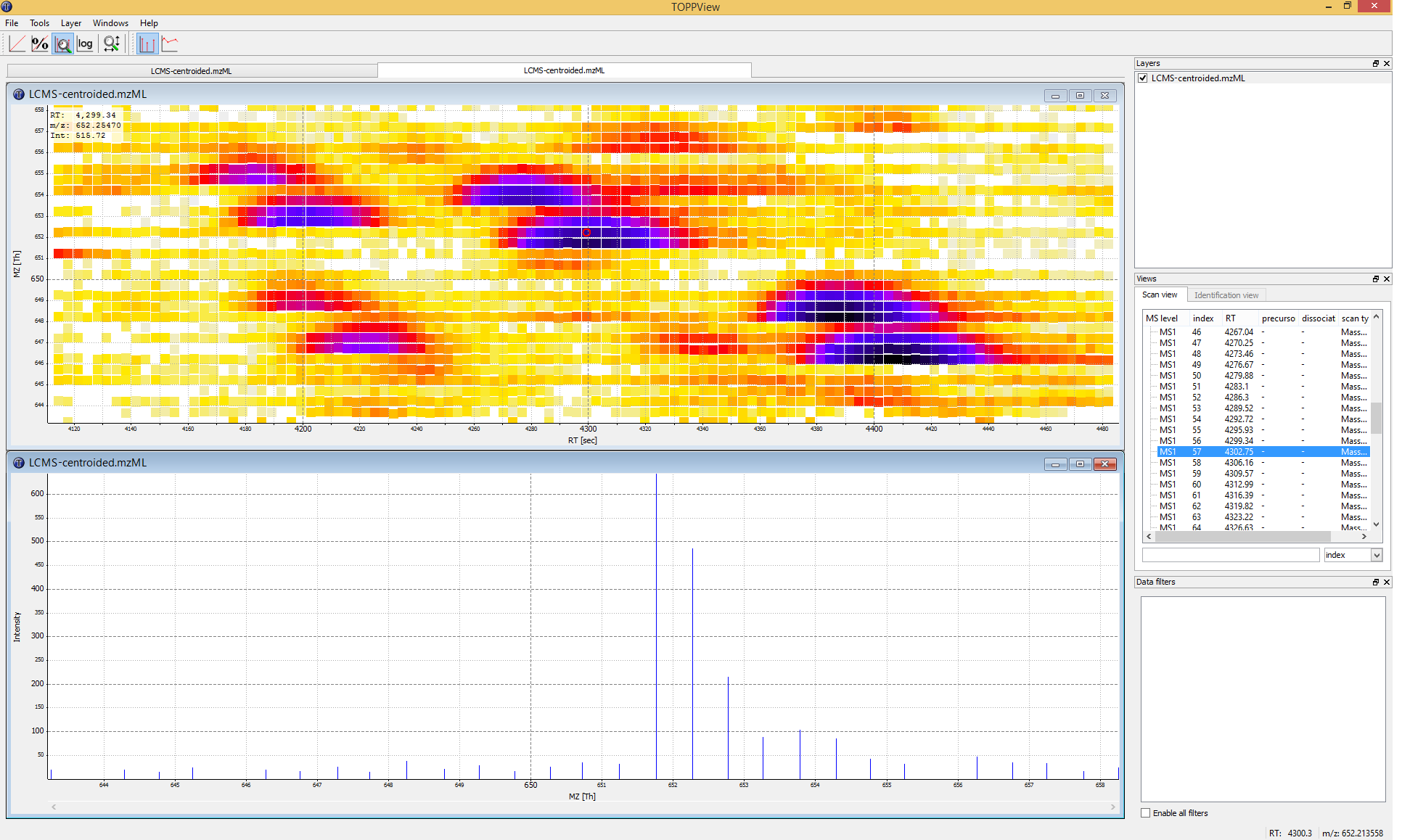
to obtain a global view on the whole LC-MS map - the default option Map view 2D
is the correct one and we can click the
. This file contains only a reduced LC-MS map 1
of a label-free proteomic platelet measurement recorded on an Orbitrap velos. The
other two mzML files contain technical replicates of this experiment. First, we want
to obtain a global view on the whole LC-MS map - the default option Map view 2D
is the correct one and we can click the  button.
button.
 key to zoom to this area or use your mouse wheel to zoom in and out.
key to zoom to this area or use your mouse wheel to zoom in and out.
 or
or  or the mouse wheel (scroll up and down).
or the mouse wheel (scroll up and down).
 zooms out to show the full LC-MS map (and also resets the zoom history).
zooms out to show the full LC-MS map (and also resets the zoom history). (shift) key.
(shift) key.
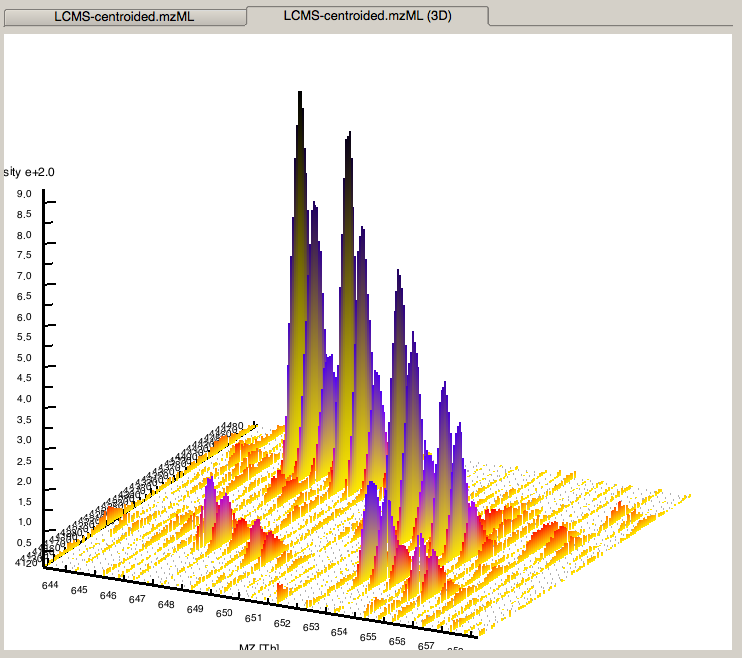
 and examine your data in 3D mode (see Fig. 4)
and examine your data in 3D mode (see Fig. 4)
Note: On macOS, due to a bug in one of the external libraries used by OpenMS, you will see a small window of the 3D mode when switching to 2D. Close the 3D tab in order to get rid of it.
 and choose a TOPP tool (e.g., FileInfo) and inspect the results.
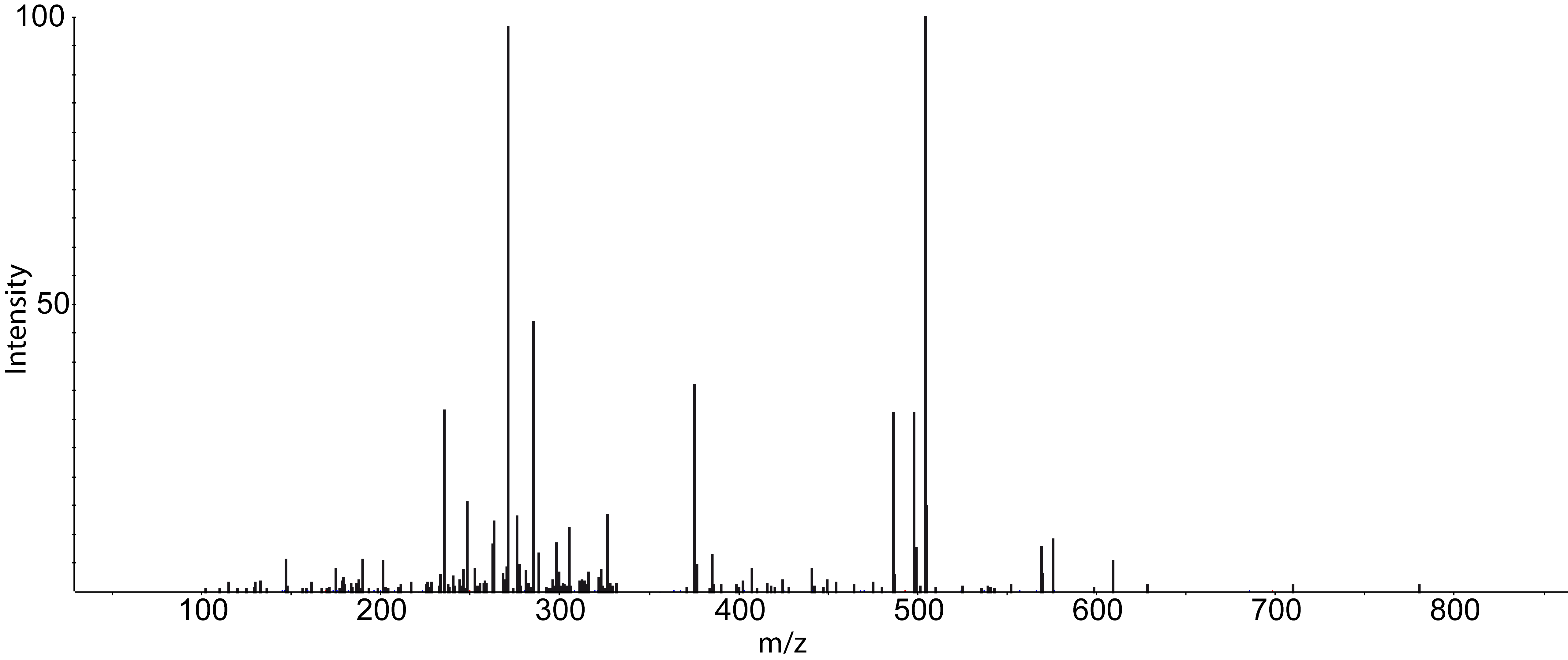
and choose a TOPP tool (e.g., FileInfo) and inspect the results.Dependent on your data MS/MS spectra can be visualized as well (see Fig.5) . You can do so,
by double-click on the MS/MS spectrum shown in scan view.
Using OpenMS in combination with KNIME, you can create, edit, open, save, and run
workflows that combine TOPP tools with the powerful data analysis capabilities of KNIME.
Workflows can be created conveniently in a graphical user interface. The parameters of all
involved tools can be edited within the application and are also saved as part of the
workflow. Furthermore, KNIME interactively performs validity checks during the
workflow editing process, in order to make it more difficult to create an invalid workflow.
Throughout most parts of this tutorial you will use KNIME to create and execute workflows.
The first step is to make yourself familiar with KNIME. Additional information on basic usage
of KNIME can be found on the KNIME Getting Started page. However, the most important
concepts will also be reviewed in this tutorial.
Before we can start with the tutorial we need to install all the required extensions for KNIME. Since KNIME 3.2.1 the program automatically detects missing plugins when you open a workflow but to make sure that the right source for the OpenMS plugin is chosen, please follow the instructions here. First, we install some additional extensions that are required by our OpenMS nodes or used in the Tutorials e.g. for visualization and file handling.

 drop-down list select
drop-down list select 
 and follow the instructions (you may but don’t need to restart KNIME now)
and follow the instructions (you may but don’t need to restart KNIME now)

 drop-down list select
drop-down list select 
 and follow the instructions and after a restart of KNIME the dependencies will be
installed.
and follow the instructions and after a restart of KNIME the dependencies will be
installed.In addition, we need to install R for the statistical downstream analysis. Choose the directory
that matches your operating system, double-click the R installer and follow the instructions. We
recommend to use the default settings whenever possible. On macOS you also need to install
XQuartz from the same directory.
Afterwards open your R installation. If you use Windows, you will find an ”R x64 3.6.X” icon
on your desktop. If you use macOS, you will find R in your Applications folder. In R type the
following lines (you might also copy them from the file  folder on the USB stick):
folder on the USB stick):
install.packages('Rserve',,"http://rforge.net/",type="source") install.packages("Cairo") install.packages("devtools") install.packages("ggplot2") install.packages("ggfortify") if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install() BiocManager::install(c("MSstats"))
In KNIME, click on  ,
select the category
,
select the category  and
set the ”Path to R Home” to your installation path. You can use the following settings, if you
installed R as described above:
and
set the ”Path to R Home” to your installation path. You can use the following settings, if you
installed R as described above:
You are now ready to install the OpenMS nodes.

We included a custom KNIME update site to install the OpenMS KNIME plugins from the USB stick. If you do not have a stick available, please see below.
 (in the upper right corner of the dialog) to define a new update site. In the opening
dialog enter the following details.
(in the upper right corner of the dialog) to define a new update site. In the opening
dialog enter the following details.  KNIME will show you all the contents of the added Update Site.
KNIME will show you all the contents of the added Update Site.
 drop-down list.
drop-down list.
 .
.
Alternatively, you can try these steps that will install the OpenMS KNIME plugins from the internet. Note that download can be slow.
 (in the upper right corner of the dialog) to define a new update site. In the opening
dialog enter the following details.
(in the upper right corner of the dialog) to define a new update site. In the opening
dialog enter the following details. 
 KNIME will show you all the contents of the added Update Site.
KNIME will show you all the contents of the added Update Site.
 drop-down list.
drop-down list.
 .
.
A workflow is a sequence of computational steps applied to a single or multiple input data to
process and analyze the data. In KNIME such workflows are implemented graphically by
connecting so-called nodes. A node represents a single analysis step in a workflow. Nodes have
input and output ports where the data enters the node or the results are provided for other
nodes after processing, respectively. KNIME distinguishes between different port types,
representing different types of data. The most common representation of data in KNIME
are tables (similar to an excel sheet). Ports that accept tables are marked with a
small triangle. For OpenMS nodes, we use a different port type, so called file ports,
representing complete files. Those ports are marked by a small blue box. Filled blue
boxes represent mandatory inputs and empty blue boxes optional inputs. The same
holds for output ports, despite you can deactivate them in the configuration dialog
(double-click on node) under the OutputTypes tab. After execution, deactivated
ports will be marked with a red cross and downstream nodes will be inactive (not
configurable).
A typical OpenMS workflow in KNIME can be divided in two conceptually different parts:
Moreover, nodes can have three different states, indicated by the small traffic light below the node.
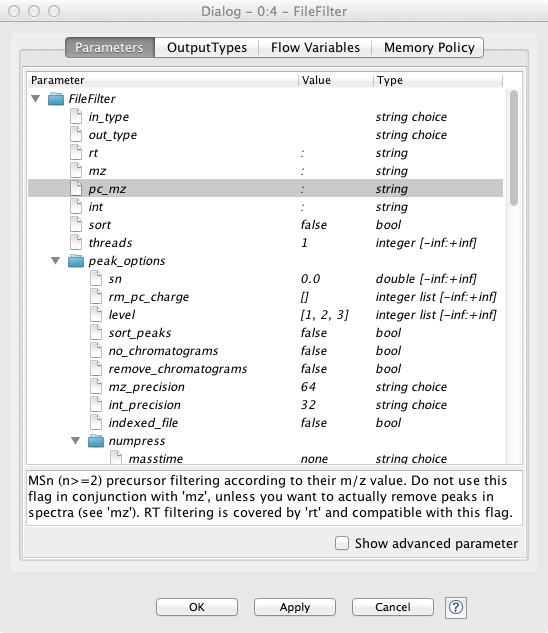
If the node execution fails, the node will switch to the red state. Other anomalies and warnings like missing information or empty results will be presented with a yellow exclamation mark above the traffic light. Most nodes will be configured as soon as all input ports are connected. Some nodes need to know about the output of the predecessor and may stay red until the predecessor was executed. If nodes still remain in a red state, probably additional parameters have to be provided in the configuration dialog that can neither be guessed from the data nor filled with sensible defaults. In this case, or if you want to customize the default configuration in general, you can open the configuration dialog of a node with a double-click on the node. For all OpenMS nodes you will see a configuration dialog like the one shown in Figure 6.
Note: OpenMS distinguishes between normal parameters and advanced parameters.
Advanced parameters are by default hidden from the users since they should only
rarely be customized. In case you want to have a look at the parameters or need to
customize them in one of the tutorials you can show them by clicking on the checkbox
 in
the lower part of the dialog. Afterwards the parameters are shown in a light gray
color.
in
the lower part of the dialog. Afterwards the parameters are shown in a light gray
color.
The dialog shows the individual parameters, their current value and type, and, in the lower part of the dialog, the documentation for the currently selected parameter. Please also note the tabs on the top of the configuration dialog. In the case of OpenMS nodes, there will be another tab called OutputTypes. It contains dropdown menus for every output port that let you select the output filetype that you want the node to return (if the tool supports it). For optional output ports you can select Inactive such that the port is crossed out after execution and the associated generation of the file and possible additional computations are not performed. Note that this will deactivate potential downstream nodes connected to this port.
The graphical user interface (GUI) of KNIME consists of different components or so-called panels that are shown in Figure 7. We will briefly introduce the individual panels and their purposes below.
 or (
or ( +)
+) depending on your OS and settings. Multiselection happens via dragging rectangles
with the mouse or adding elements to the selection by clicking them while holding
down
depending on your OS and settings. Multiselection happens via dragging rectangles
with the mouse or adding elements to the selection by clicking them while holding
down  .
.
 from the appearing context menu. Remember to save your workflow often with the
from the appearing context menu. Remember to save your workflow often with the
 +
+ shortcut.
shortcut.
 . Nodes for managing files (e.g., Input Files or Output Folders) can be found in
. Nodes for managing files (e.g., Input Files or Output Folders) can be found in  . You can search the node repository by typing the node name into the small text
box in the upper part of the node repository.
. You can search the node repository by typing the node name into the small text
box in the upper part of the node repository.
Workflows can easily be created by a right click in the Workflow Explorer followed by clicking
on  .
.
To be able to share a workflow with others, KNIME supports the import and export of
complete workflows. To export a workflow, select it in the Workflow Explorer and select
 .
KNIME will export workflows as a knwf file containing all the information on nodes, their
connections, and their parameter configuration. Those knwf files can again be imported by
selecting
.
KNIME will export workflows as a knwf file containing all the information on nodes, their
connections, and their parameter configuration. Those knwf files can again be imported by
selecting  .
.
Note: For your convenience we added all workflows discussed in this tutorial to the
 folder on the USB Stick. Additionally, the workflow files can be found on our GitHub
repository. If you want to check your own workflow by comparing it to the solution or got stuck,
simply import the full workflow from the corresponding knwf file and after that double-click it
in your KNIME Workflow repository to open it.
folder on the USB Stick. Additionally, the workflow files can be found on our GitHub
repository. If you want to check your own workflow by comparing it to the solution or got stuck,
simply import the full workflow from the corresponding knwf file and after that double-click it
in your KNIME Workflow repository to open it.
In this tutorial, a lot of the workflows will be created based on the workflow from a previous task. To keep the intermediate workflows, we suggest you create copies of your workflows so you can see the progress. To create a copy of your workflow, save it, close it and follow the next steps.
 .
.
 .
.
 .
.Note: To rename a workflow it has to be closed, too.
Let us now start with the creation of our very first, very simple workflow. As a first step, we will gather some basic information about the data set before starting the actual development of a data analysis workflow. This minimal workflow can also be used to check if all requirements are met and that your system is compatible.
 and a FileInfo node (to be found in the category
and a FileInfo node (to be found in the category  ) to the workflow.
) to the workflow.
Note: In case you are unsure about which node port to use, hovering the cursor over the port in question will display the port name and what kind of input it expects.
The complete workflow is shown in Figure 8. FileInfo can produce two different kinds of output files.
 . In the file system browser select
. In the file system browser select  and click
and click  . Afterwards close the dialog by clicking
. Afterwards close the dialog by clicking  .
.Note: Make sure to use the “tiny” version this time, not “small”, for the sake of faster workflow execution.
 to select an output directory for the generated data.
to select an output directory for the generated data.
 (shift key + F7; or the button with multiple green triangles in the KNIME Toolbar) to
execute the complete workflow. You can also right click on any node of your workflow and
select
(shift key + F7; or the button with multiple green triangles in the KNIME Toolbar) to
execute the complete workflow. You can also right click on any node of your workflow and
select  from the context menu.
from the context menu.
 . You can then open the text file and inspect its contents. You will find some basic
information of the data contained in the mzML file, e.g., the total number of spectra and
peaks, the RT and m/z range, and how many MS1 and MS2 spectra the file
contains.
. You can then open the text file and inspect its contents. You will find some basic
information of the data contained in the mzML file, e.g., the total number of spectra and
peaks, the RT and m/z range, and how many MS1 and MS2 spectra the file
contains.Workflows are typically constructed to process a large number of files automatically. As a simple example, consider you would like to convert multiple Thermo Raw files into the mzML format. We will now modify the workflow to compute the same information on three different files and then write the output files to a folder.
 .
.
 . In the filesystem browser we select all three files from the directory
. In the filesystem browser we select all three files from the directory  . And close the dialog with
. And close the dialog with  .
.
 .
.
In case you had trouble to understand what ZipLoopStart and ZipLoopEnd do - here is a brief explanation:
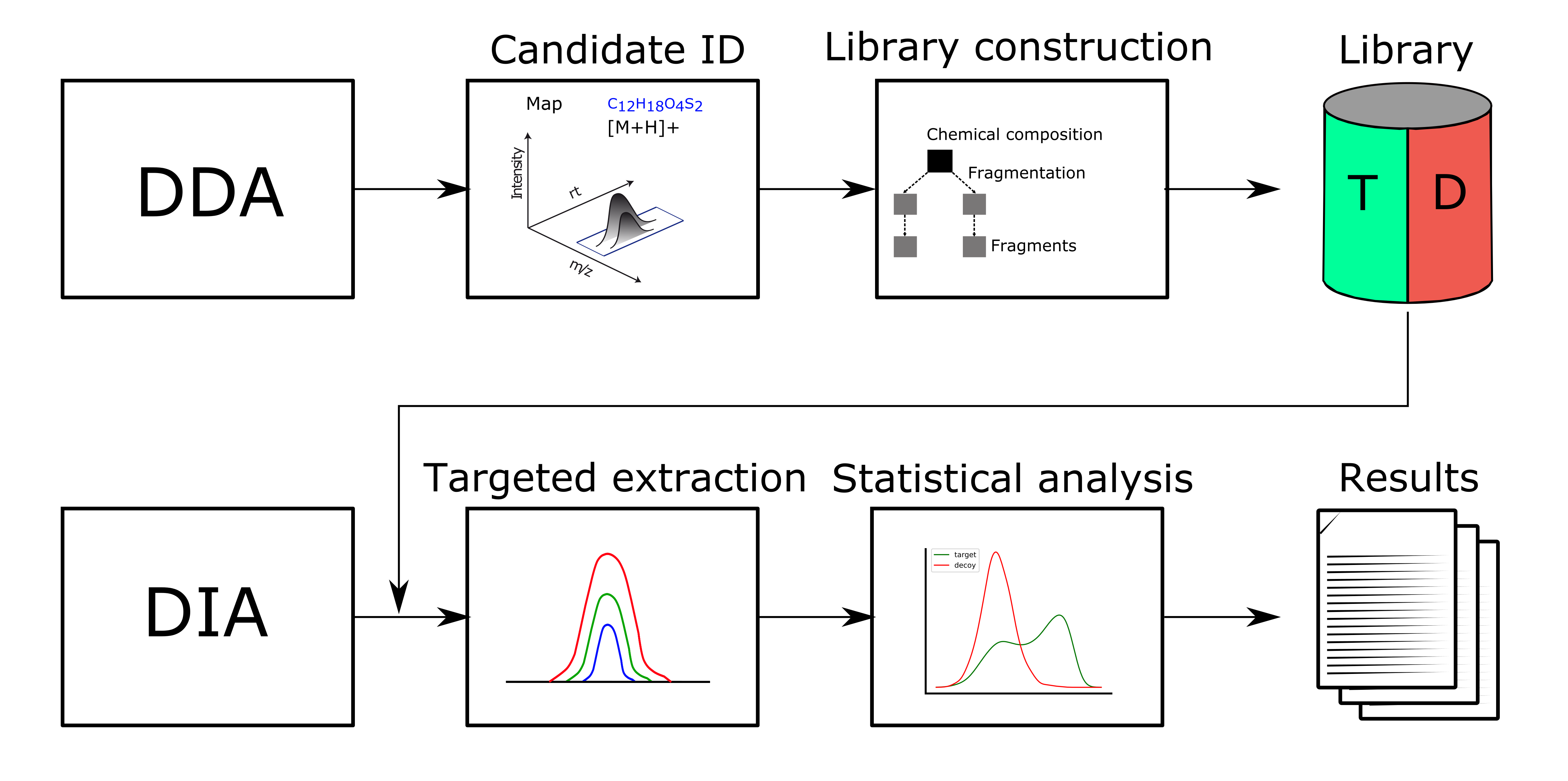
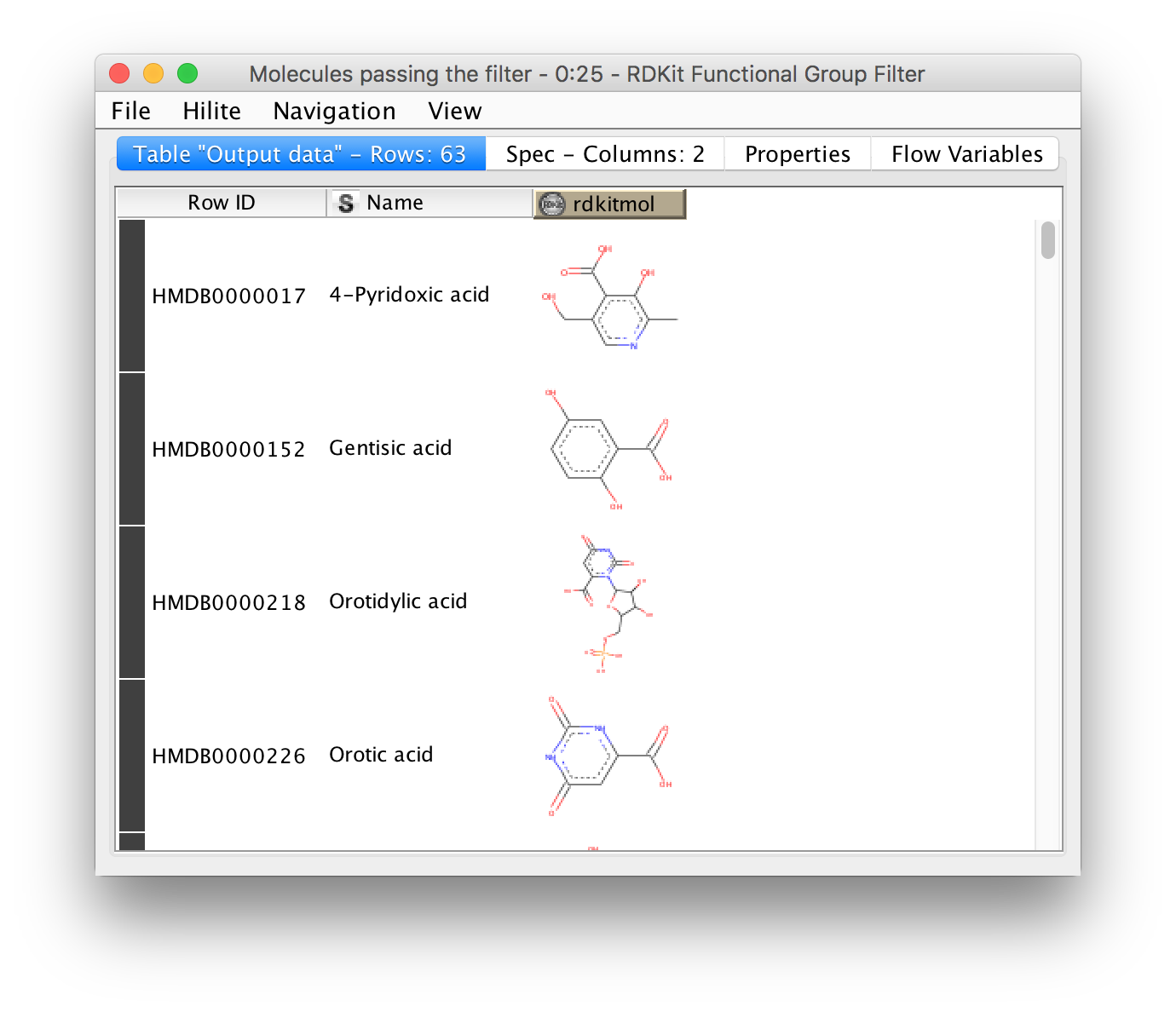
Metabolomics analyses often involve working with chemical structures. Popular cheminformatic toolkits such as RDKit [7] or CDK [8] are available as KNIME plugins and allow us to work with chemical structures directly from within KNIME. In particular, we will use KNIME and RDKit to visualize a list of compounds and filter them by predefined substructures. Chemical structures are often represented as SMILES (Simplified molecular input line entry specification), a simple and compact way to describe complex chemical structures as text. For example, the chemical structure of L-alanine can be written as the SMILES string C[C@H](N)C(O)=O. As we will discuss later, all OpenMS tools that perform metabolite identification will report SMILES as part of their result, which can then be further processed and visualized using RDKit and KNIME.
Perform the following steps to build the workflow shown in in Fig. 10. You will use this workflow to visualize a list of SMILES strings and filter them by predefined substructures:
 . This file has been exported from the Human Metabolome Database (HMDB)
and contains the portion of the human metabolome that has been detected and
quantified. The file preview on the bottom of the dialog shows that each compound
is given by its HMDB accession, compound name, and SMILES string. Click on the
column header ’SMILES’ to change its properties. Change the column type from
’string’ to ’smiles’ and close the dialog with
. This file has been exported from the Human Metabolome Database (HMDB)
and contains the portion of the human metabolome that has been detected and
quantified. The file preview on the bottom of the dialog shows that each compound
is given by its HMDB accession, compound name, and SMILES string. Click on the
column header ’SMILES’ to change its properties. Change the column type from
’string’ to ’smiles’ and close the dialog with  . Afterwards the SMILES column will be visualized as chemical structures instead
of text directly within all KNIME tables.
. Afterwards the SMILES column will be visualized as chemical structures instead
of text directly within all KNIME tables.
Workflows can get rather complex and may contain dozens or even hundreds of nodes. KNIME provides a simple way to improve handling and clarity of large workflows:
Metanodes allow to bundle several nodes into a single Metanode.
Task
Select multiple nodes (e.g. all nodes of the ZipLoop including the start and end node). To select
a set of nodes, draw a rectangle around them with the left mouse button or hold
 to
add/remove single nodes from the selection. Pro-tip: There is a
to
add/remove single nodes from the selection. Pro-tip: There is a  option when you right-click a node in a loop, that does exactly that for you. Then, open the
context menu (right-click on a node in the selection) and select
option when you right-click a node in a loop, that does exactly that for you. Then, open the
context menu (right-click on a node in the selection) and select  .
Enter a caption for the Metanode. The previously selected nodes are now contained in the
Metanode. Double-clicking on the Metanode will display the contained nodes in a new tab
window.
.
Enter a caption for the Metanode. The previously selected nodes are now contained in the
Metanode. Double-clicking on the Metanode will display the contained nodes in a new tab
window.
Task
Create the Metanode to let it behave like an encapsulated single node. First select the
Metanode, open the context menu (right-click) and select  . The
differences between Metanodes and their wrapped counterparts are marginal (and only apply
when exposing user inputs and workflow variables). Therefore we suggest to use standard
Metanodes to clean up your workflow and cluster common subparts until you actually notice
their limits.
. The
differences between Metanodes and their wrapped counterparts are marginal (and only apply
when exposing user inputs and workflow variables). Therefore we suggest to use standard
Metanodes to clean up your workflow and cluster common subparts until you actually notice
their limits.
Task
Undo the packaging. First select the (Wrapped) Metanode, open the context menu (right-click)
and select  .
.
KNIME provides a large number of nodes for a wide range of statistical analysis, machine learning, data processing, and visualization. Still, more recent statistical analysis methods, specialized visualizations or cutting edge algorithms may not be covered in KNIME. In order to expand its capabilities beyond the readily available nodes, external scripting languages can be integrated. In this tutorial, we primarily use scripts of the powerful statistical computing language R. Note that this part is considered advanced and might be difficult to follow if you are not familiar with R. In this case you might skip this part.
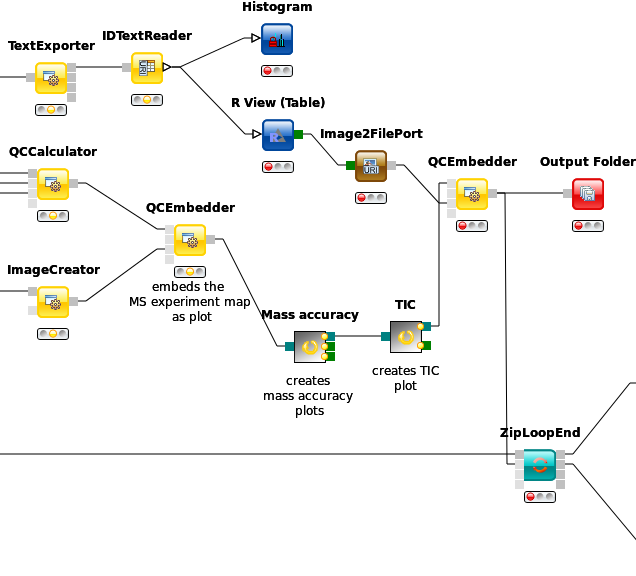
R View (Table) allows to seamlessly include R scripts into KNIME. We will demonstrate on a minimal example how such a script is integrated.
Task
First we need some example data in KNIME, which we will generate using the Data Generator
node. You can keep the default settings and execute the node. The table contains four columns,
each containing random coordinates and one column containing a cluster number (Cluster_0 to
Cluster_3). Now place a R View (Table) node into the workflow and connect the
upper output port of the Data Generator node to the input of the R View (Table)
node. Right-click and configure the node. If you get an error message like ”Execute
failed: R_HOME does not contain a folder with name ’bin’.” or ”Execution failed: R
Home is invalid.”: please change the R settings in the preferences. To do so open
 and
enter the path to your R installation (the folder that contains the bin directory (e.g.,
and
enter the path to your R installation (the folder that contains the bin directory (e.g.,
 ).
).
If you get an error message like: ”Execute failed: Could not find Rserve package. Please install it
in your R installation by running
”install.packages(’Rserve’)”.” You may need to run your R binary as administrator (In windows
explorer: right-click ”Run as administrator”) and enter install.packages(’Rserve’) to install the
package.
If R is correctly recognized we can start writing an R script. Consider that we are interested in
plotting the first and second coordinates and color them according to their cluster number. In R
this can be done in a single line. In the R View (Table) text editor, enter the following code:
plot(x=knime.in$Universe_0_0, y=knime.in$Universe_0_1, main="Plotting column Universe_0_0 vs. Universe_0_1", col=knime.in$"Cluster Membership")
Explanation: The table provided as input to the R View (Table) node is available as R data.frame with name knime.in. Columns (also listed on the left side of the R View window) can be accessed in the usual R way by first specifying the data.frame name and then the column name (e.g. knime.in$Universe_0_0). plot is the plotting function we use to generate the image. We tell it to use the data in column Universe_0_0 of the dataframe object knime.in (denoted as knime.in$Universe_0_0) as x-coordinate and the other column knime.in$Universe_0_1 as y-coordinate in the plot. main is simply the main title of the plot and col the column that is used to determine the color (in this case it is the Cluster Membership column).
Now press the  and
and
 buttons.
buttons.
Note: Note that we needed to put some extra quotes around Cluster Membership. If we omit those, R would interpret the column name only up to the first space (knime.in$Cluster) which is not present in the table and leads to an error. Quotes are regularly needed if column names contain spaces, tabs or other special characters like $ itself.
In this chapter, we will build a workflow with OpenMS / KNIME to quantify a label-free experiment. Label-free quantification is a method aiming to compare the relative amounts of proteins or peptides in two or more samples. We will start from the minimal workflow of the last chapter and, step-by-step, build a label-free quantification workflow.
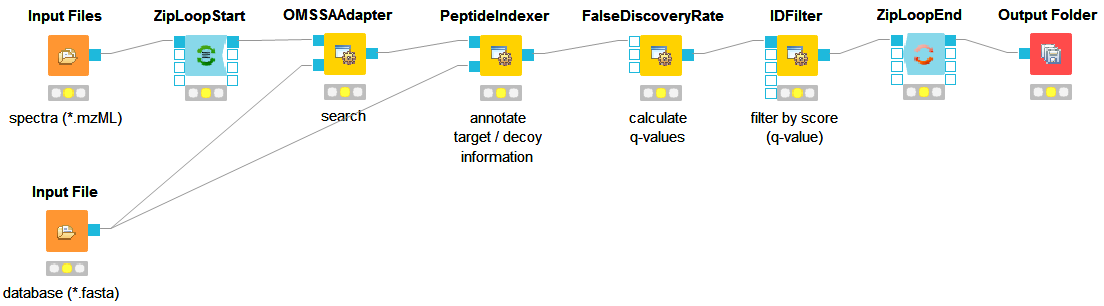
As a start, we will extend the minimal workflow so that it performs a peptide identification using the OMSSA [9] search engine. Since OpenMS version 1.10, OMSSA is included in the OpenMS installation, so you do not need to download and install it yourself.
 . This is a reduced toy dataset where each of the three runs contains a constant
background of S. pyogenes peptides as well as human spike-in peptides in different
concentrations. [10]
. This is a reduced toy dataset where each of the three runs contains a constant
background of S. pyogenes peptides as well as human spike-in peptides in different
concentrations. [10]
 , and we are almost done. Just make sure you have connected the ZipLoopStart
node with the in port of the OMSSAAdapter node.
, and we are almost done. Just make sure you have connected the ZipLoopStart
node with the in port of the OMSSAAdapter node.
 , and we have a very basic peptide identification workflow.
, and we have a very basic peptide identification workflow.Note: You might also want to save your new identification workflow under a different name. Have a look at Section 2.4.6 for information on how to create copies of workflows.
 from the menu and select the idXML file that OMSSAAdapter generated (it
is located within the output directory that you specified when starting the
pipeline).
from the menu and select the idXML file that OMSSAAdapter generated (it
is located within the output directory that you specified when starting the
pipeline).
 . Using this view, you can see all identified peptides and browse the corresponding MS2
spectra.
. Using this view, you can see all identified peptides and browse the corresponding MS2
spectra.Note: Opening the output file of OMSSAAdapter (the idXML file) directly is also possible, but the direct visualization of an idXML file is less useful.
 to your workflow and connect the output port of your OMSSAAdapter (the same port
your ZipLoopEnd is connected to) to its input port. This tool will convert the
idXML file to a more human-readable text file which can also be read into
a KNIME table using the IDTextReader node. Add an IDTextReader node
(
to your workflow and connect the output port of your OMSSAAdapter (the same port
your ZipLoopEnd is connected to) to its input port. This tool will convert the
idXML file to a more human-readable text file which can also be read into
a KNIME table using the IDTextReader node. Add an IDTextReader node
( ) after TextExporter and execute it. Now you can right-click IDTextReader and select
) after TextExporter and execute it. Now you can right-click IDTextReader and select
 to browse your peptide identifications.
to browse your peptide identifications.
 ) node after IDTextReader, double-click it, select peptide_charge as Histogram column, hit
) node after IDTextReader, double-click it, select peptide_charge as Histogram column, hit
 , and execute it. Right-clicking and selecting
, and execute it. Right-clicking and selecting  will open a plot showing the charge state distribution of your identifications.
will open a plot showing the charge state distribution of your identifications.In the next step, we will tweak the parameters of OMSSA to better reflect the instrument’s accuracy. Also, we will extend our pipeline with a false discovery rate (FDR) filter to retain only those identifications that will yield an FDR of < 1 %.
Note: Whenever you change the configuration of a node, the node as well as all its successors will be reset to the Configured state (all node results are discarded and need to be recalculated by executing the nodes again).
Note: To add a modification click on the empty value field in the configuration dialog to
open the list editor dialog. In the new dialog click  . Then select the newly added modification to open the drop down list where you can
select the correct modification.
. Then select the newly added modification to open the drop down list where you can
select the correct modification.
 . This node needs the idXML as input as well as the database file (see Fig
12).
. This node needs the idXML as input as well as the database file (see Fig
12).Note: You can direct the files of an Input File node to more than just one destination port.
 ).
).
 . Configuring its parameter score →pep to 0.01 will do the trick. The FDR calculations
(embedded in the idXML) from the FalseDiscoveryRate node will go into the in port of
the IDFilter node.
. Configuring its parameter score →pep to 0.01 will do the trick. The FDR calculations
(embedded in the idXML) from the FalseDiscoveryRate node will go into the in port of
the IDFilter node.
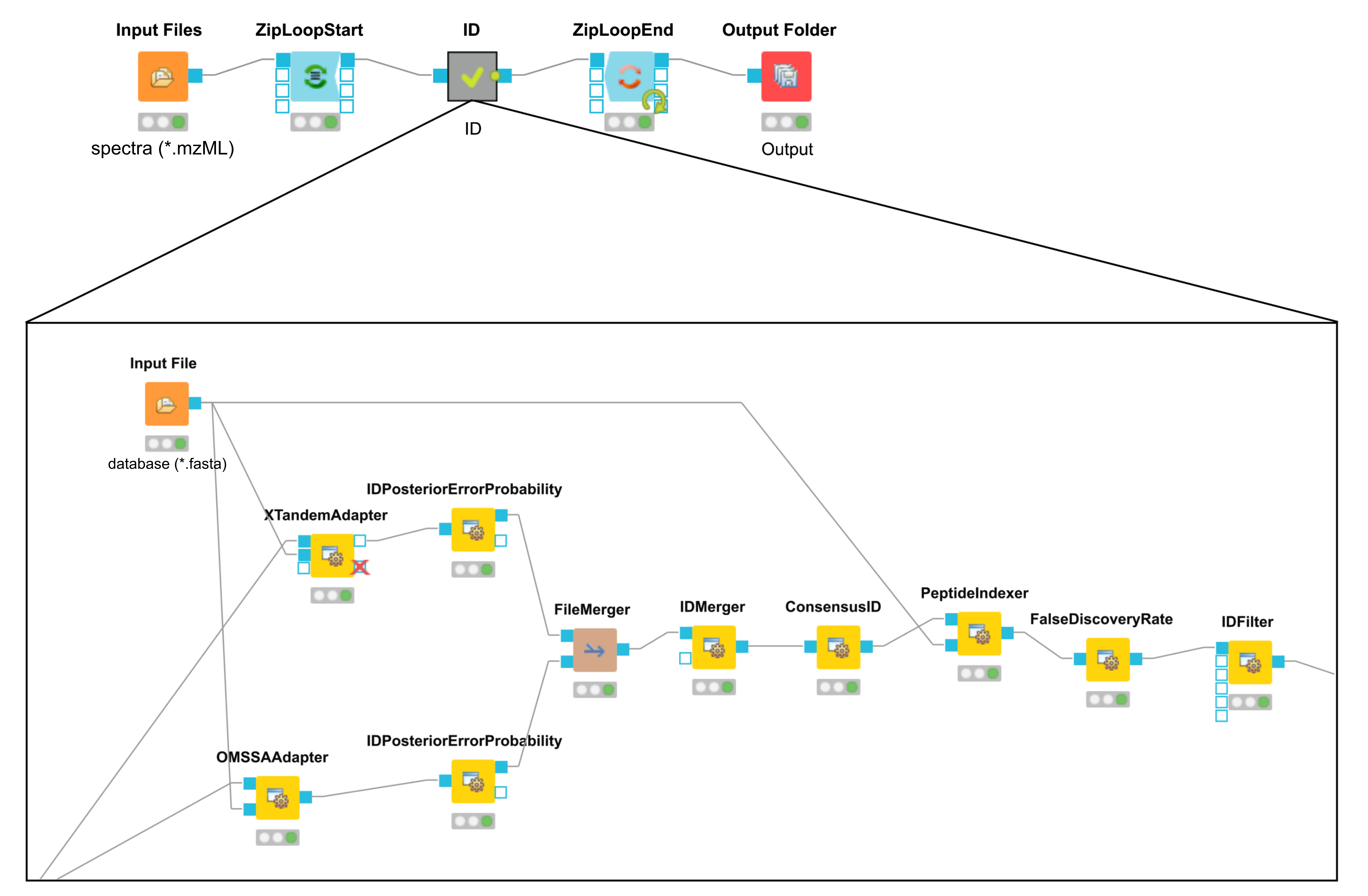
Note: The finished identification workflow is now sufficiently complex that we might
want to encapsulate it in a Metanode. For this, select all nodes inside the ZipLoop
(including the Input File node) and right-click to select  and name it ID. Metanodes are useful when you construct even larger workflows and
want to keep an overview.
and name it ID. Metanodes are useful when you construct even larger workflows and
want to keep an overview.
Note: If you are ahead of the tutorial or later on, you can further improve your FDR identification workflow by a so-called consensus identification using several search engines. Otherwise, just continue with section 3.3.
It has become widely accepted that the parallel usage of different search engines can increase peptide identification rates in shotgun proteomics experiments. The ConsensusID algorithm is based on the calculation of posterior error probabilities (PEP) and a combination of the normalized scores by considering missing peptide sequences.
 node and set its parameters and ports analogously to the OMSSAAdapter. In XTandem,
to get more evenly distributed scores, we decrease the number of candidates a bit
by setting the precursor mass tolerance to 5 ppm and the fragment mass tolerance
to 0.1 Da.
node and set its parameters and ports analogously to the OMSSAAdapter. In XTandem,
to get more evenly distributed scores, we decrease the number of candidates a bit
by setting the precursor mass tolerance to 5 ppm and the fragment mass tolerance
to 0.1 Da.
 node to the output of each ID engine adapter node. This will calculate the PEP to
each hit and output an updated idXML.
node to the output of each ID engine adapter node. This will calculate the PEP to
each hit and output an updated idXML.
 so we can then merge the corresponding IDs with a IDMerger
so we can then merge the corresponding IDs with a IDMerger  .
.
 node. We can connect this to the PeptideIndexer and go along with our existing FDR
filtering.
node. We can connect this to the PeptideIndexer and go along with our existing FDR
filtering.Note: By default, X!Tandem takes additional enzyme cutting rules into consideration (besides the specified tryptic digest). Thus for the tutorial files, you have to set PeptideIndexer’s enzyme →specificity parameter to none to accept X!Tandems non-tryptic identifications as well.
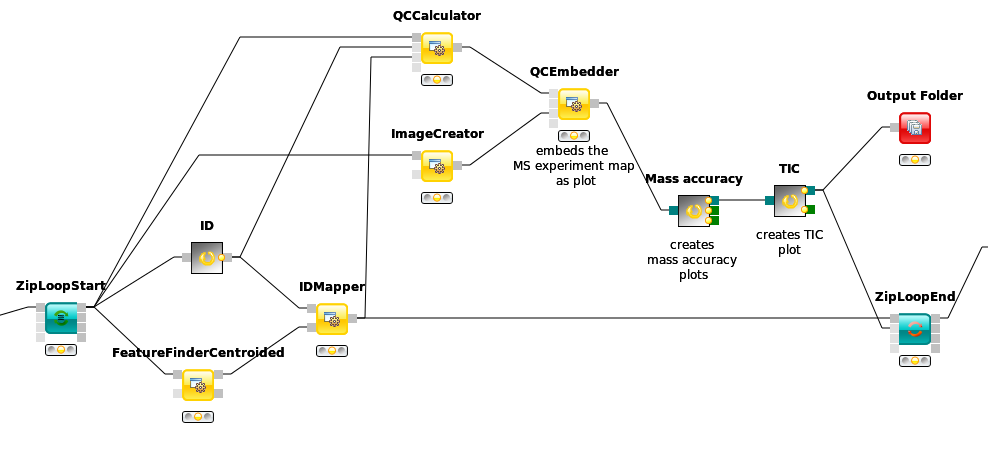
In the end the ID processing part of the workflow can be collapsed into a Metanode to keep the structure clean (see Figure 13).
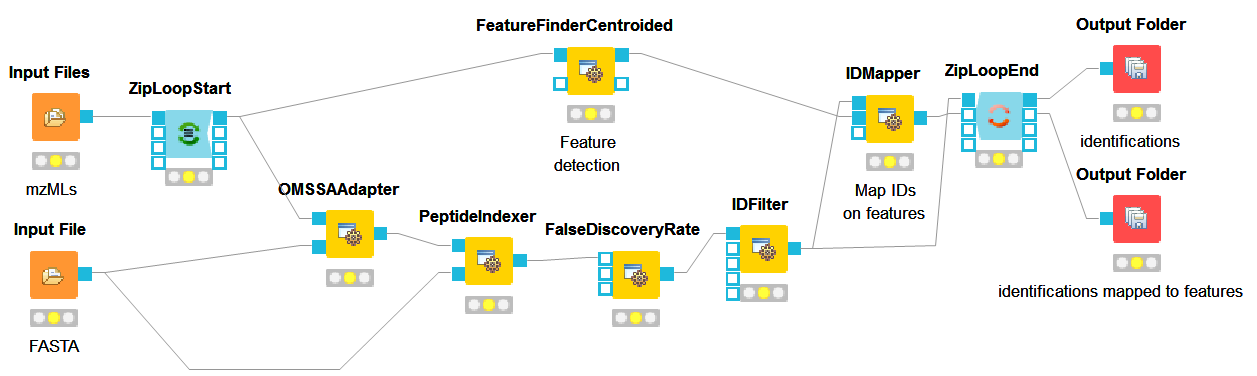
Now that we have successfully constructed a peptide identification pipeline, we can add quantification capabilities to our workflow.
 which gets input from the first output port of the ZipLoopStart node. Also, add
an IDMapper node (from
which gets input from the first output port of the ZipLoopStart node. Also, add
an IDMapper node (from  ) which receives input from the FeatureFinderCentroided node (Port 1) and the
ID Metanode (or IDFilter node (Port 0) if you haven’t used the Metanode). The
output of the IDMapper is then connected to an in port of the ZipLoopEnd node.
) which receives input from the FeatureFinderCentroided node (Port 1) and the
ID Metanode (or IDFilter node (Port 0) if you haven’t used the Metanode). The
output of the IDMapper is then connected to an in port of the ZipLoopEnd node.
 and add it to a new layer (
and add it to a new layer ( ). The features are now visualized on top of your raw data. If you zoom in on a small
region, you should be able to see the individual boxes around features that have
been detected (see Fig. 14). If you hover over the the feature centroid (small circle
indicating the chromatographic apex of monoisotopic trace) additional information
of the feature is displayed.
). The features are now visualized on top of your raw data. If you zoom in on a small
region, you should be able to see the individual boxes around features that have
been detected (see Fig. 14). If you hover over the the feature centroid (small circle
indicating the chromatographic apex of monoisotopic trace) additional information
of the feature is displayed.
Note: The chromatographic RT range of a feature is about 30-60 s and its m/z range
around 2.5 m/z in this dataset. If you have trouble zooming in on a feature, select
the full RT range and zoom only into the m/z dimension by holding down
 (
( on macOS) and repeatedly dragging a narrow box from the very left to the very
right.
on macOS) and repeatedly dragging a narrow box from the very left to the very
right.
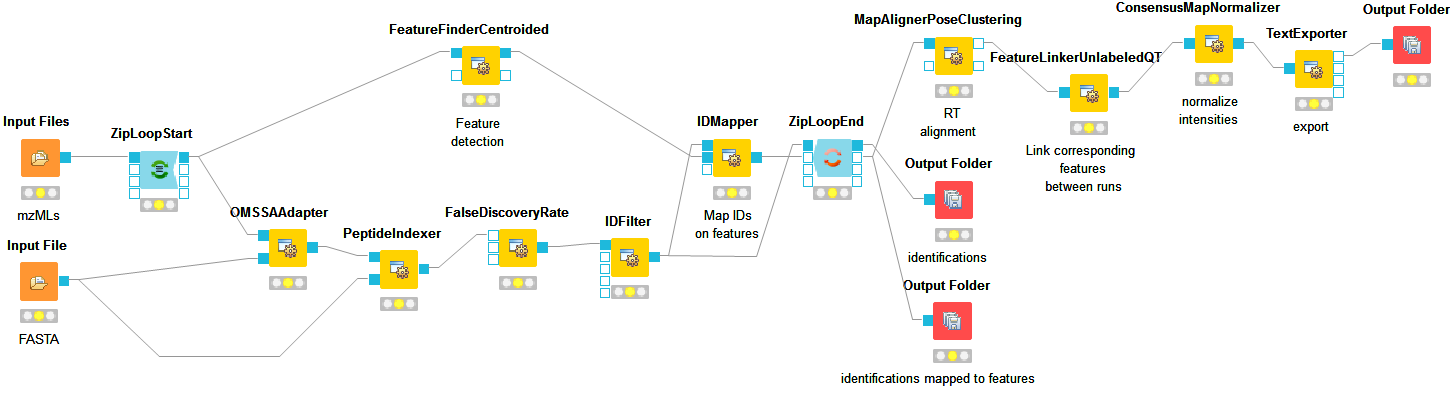
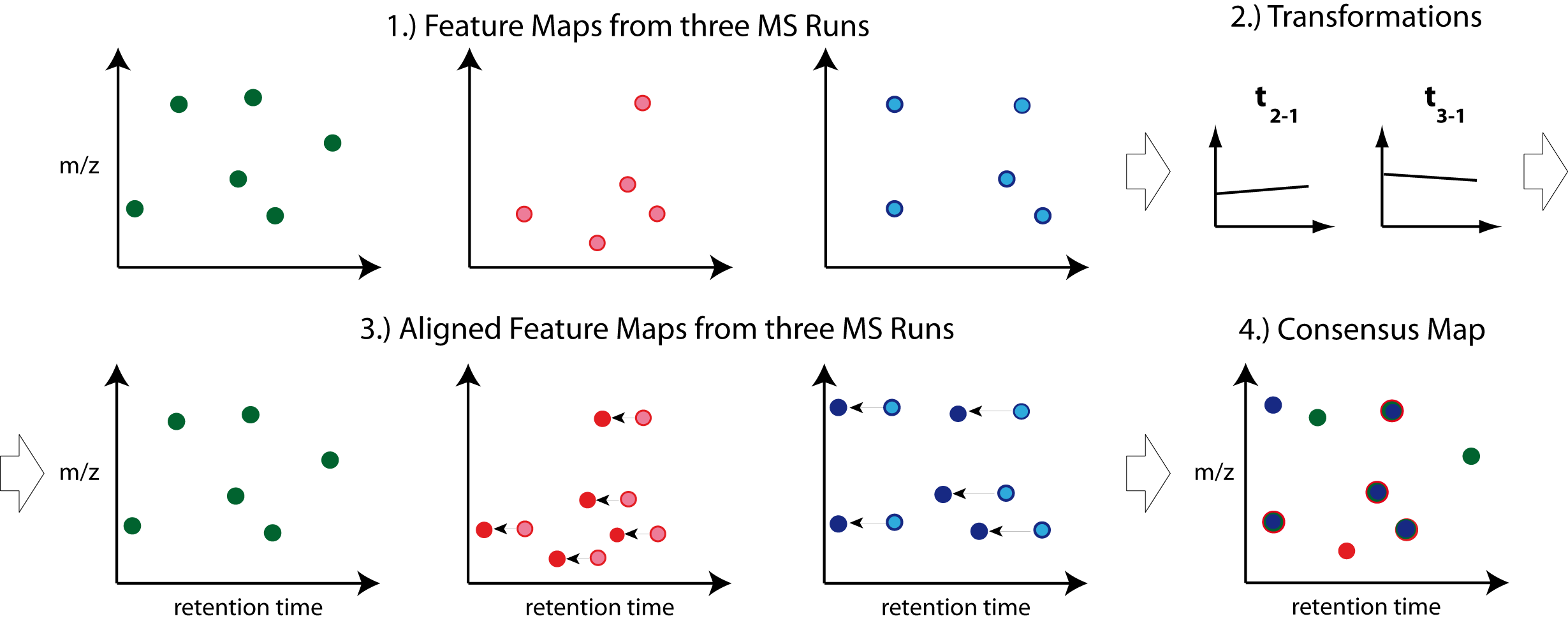
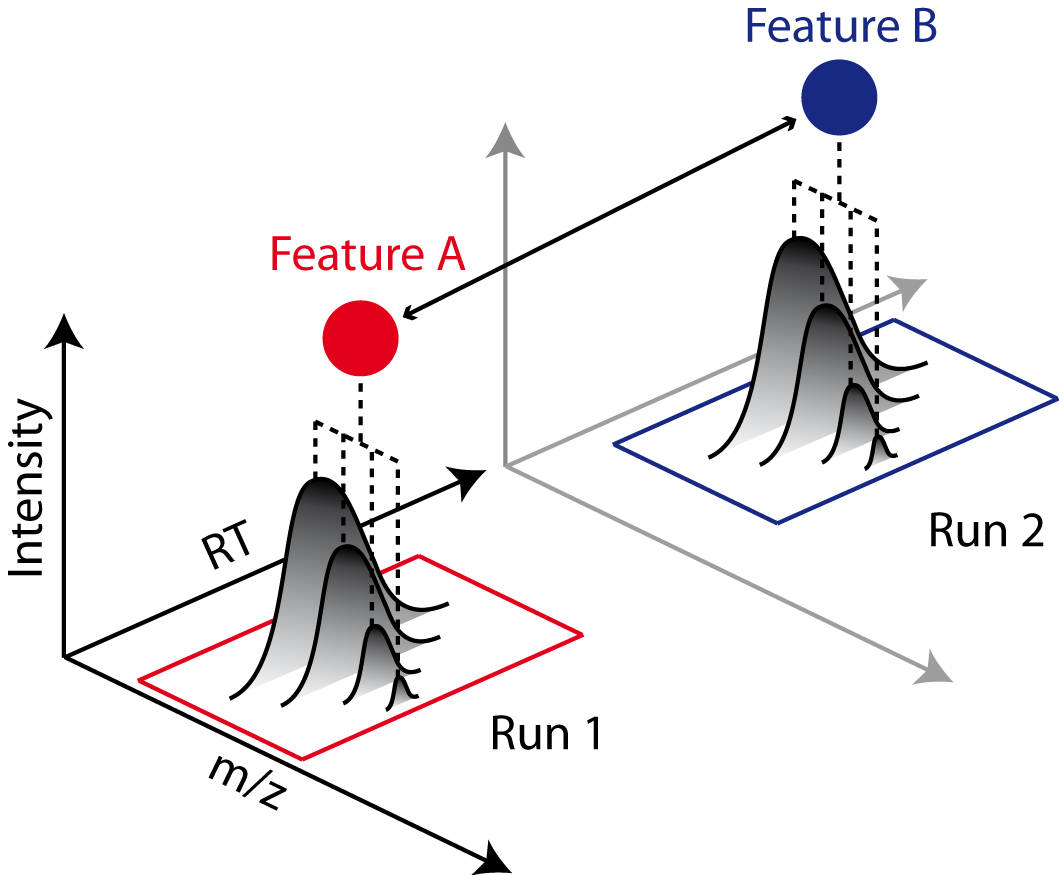
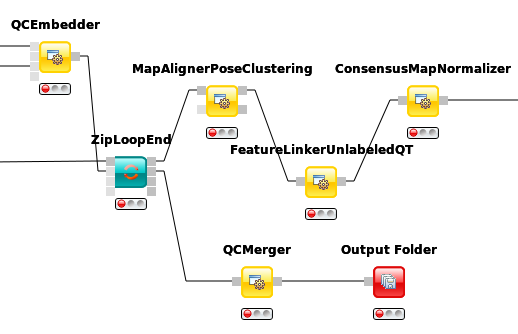
So far, we successfully performed peptide identification as well as quantification on individual LC-MS runs. For differential label-free analyses, however, we need to identify and quantify corresponding signals in different experiments and link them together to compare their intensities. Thus, we will now run our pipeline on all three available input files and extend it a bit further, so that it is able to find and link features across several runs.
 , we can align corresponding peptide signals to each other as closely as possible by
applying a transformation in the RT dimension.
, we can align corresponding peptide signals to each other as closely as possible by
applying a transformation in the RT dimension.Note: MapAlignerPoseClustering consumes several featureXML files and its output should still be several featureXML files containing the same features, but with the transformed RT values. In its configuration dialog, make sure that OutputTypes is set to featureXML.
 , we can then perform the actual linking of corresponding features. Its output is a
consensusXML file containing linked groups of corresponding features across the different
experiments.
, we can then perform the actual linking of corresponding features. Its output is a
consensusXML file containing linked groups of corresponding features across the different
experiments.
 as a last processing step. Configure its parameters with setting algorithm_type to median.
It will then normalize the maps in such a way that the median intensity of all input maps
is equal.
as a last processing step. Configure its parameters with setting algorithm_type to median.
It will then normalize the maps in such a way that the median intensity of all input maps
is equal.
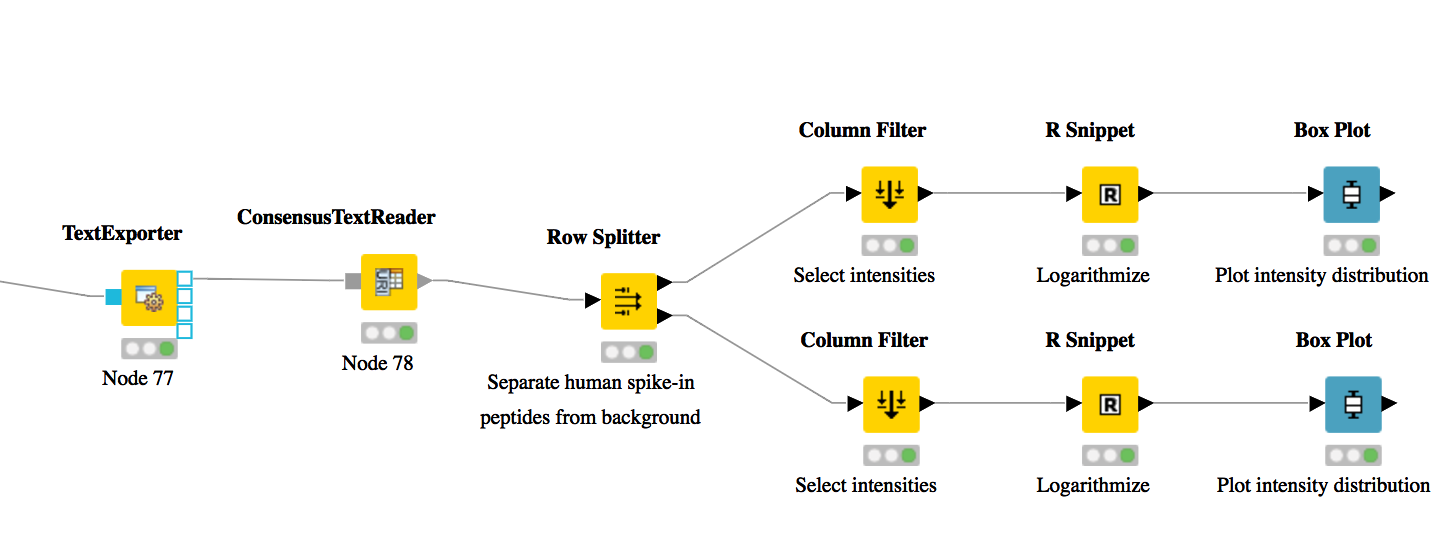
Note: You can specify the desired column separation character in the parameter settings (by default, it is set to “ ” (a space)). The output file of TextExporter can also be opened with external tools, e.g., Microsoft Excel, for downstream statistical analyses.
For downstream analysis of the quantification results within the KNIME environment, you can
use the ConsensusTextReader node in  instead of the Output Folder node to convert the output into a KNIME table (indicated by a
triangle as output port). After running the node you can view the KNIME table by
right-clicking on the ConsensusTextReader and selecting
instead of the Output Folder node to convert the output into a KNIME table (indicated by a
triangle as output port). After running the node you can view the KNIME table by
right-clicking on the ConsensusTextReader and selecting  .
Every row in this table corresponds to a so-called consensus feature, i.e., a peptide signal
quantified across several runs. The first couple of columns describe the consensus feature as a
whole (average RT and m/z across the maps, charge, etc.). The remaining columns describe the
exact positions and intensities of the quantified features separately for all input samples (e.g.,
intensity_0 is the intensity of the feature in the first input file). The last 11 columns contain
information on peptide identification.
.
Every row in this table corresponds to a so-called consensus feature, i.e., a peptide signal
quantified across several runs. The first couple of columns describe the consensus feature as a
whole (average RT and m/z across the maps, charge, etc.). The remaining columns describe the
exact positions and intensities of the quantified features separately for all input samples (e.g.,
intensity_0 is the intensity of the feature in the first input file). The last 11 columns contain
information on peptide identification.
 ) after ConsensusTextReader. Double-click it to configure. The human spike-in
peptides have accessions starting with “hum”. Thus, set the column to apply the
test to: accessions, select pattern matching as matching criterion, enter hum* into
the corresponding text field, and check the contains wild cards box. Press OK and
execute the node.
) after ConsensusTextReader. Double-click it to configure. The human spike-in
peptides have accessions starting with “hum”. Thus, set the column to apply the
test to: accessions, select pattern matching as matching criterion, enter hum* into
the corresponding text field, and check the contains wild cards box. Press OK and
execute the node.
 , connect its input port to the Filtered output port of the Row Filter, and open
its configuration dialog. We could either manually select the columns we want to
keep, or, more elegantly, select Wildcard/Regex Selection and enter intensity_? as
the pattern. KNIME will interactively show you which columns your pattern applies
to while you’re typing.
, connect its input port to the Filtered output port of the Row Filter, and open
its configuration dialog. We could either manually select the columns we want to
keep, or, more elegantly, select Wildcard/Regex Selection and enter intensity_? as
the pattern. KNIME will interactively show you which columns your pattern applies
to while you’re typing.
 after Column Filter and double-click to configure. In the R Script text editor, enter the
following code:
after Column Filter and double-click to configure. In the R Script text editor, enter the
following code:
 after the R Snippet node, execute it, and open its view. If everything went well, you
should see a significant fold change of your human peptide intensities across the three
runs.
after the R Snippet node, execute it, and open its view. If everything went well, you
should see a significant fold change of your human peptide intensities across the three
runs.
Advanced downstream data analysis of quantitative mass spectrometry-based proteomics data can be performed using MSstats [11]. This tool can be combined with an OpenMS preprocessing pipeline (e.g. in KNIME). The OpenMS experimental design is used to present the data in an MSstats-conformant way for the analysis. Here, we give an example how to utilize these resources when working with quantitative label-free data. We describe how to use OpenMS and MSstats for the analysis of the ABRF iPRG2015 dataset [12].
Note: Reanalysing the full dataset from scratch would take too long. In this tutorial session, we will focus on just the conversion process and the downstream analysis.
The R package MSstats can be used for statistical relative quantification of proteins and peptides in mass spectrometry-based proteomics. Supported are label-free as well as labeled experiments in combination with data-dependent, targeted and data-independent acquisition. Inputs can be identified and quantified entities (peptides or proteins) and the output is a list of differentially abundant entities, or summaries of their relative abundance. It depends on accurate feature detection, identification and quantification which can be performed e.g. by an OpenMS workflow.
In general MSstats can be used for data processing & visualization, as well as statistical modeling & inference. Please see [11] and http://msstats.org for further information.
The iPRG (Proteome Informatics Research Group) dataset from the study in 2015, as described in [12], aims at evaluating the effect of statistical analysis software on the accuracy of results on a proteomics label-free quantification experiment. The data is based on four artificial samples with known composition (background: 200 ng S. cerevisiae). These were spiked with different quantities of individual digested proteins, whose identifiers were masked for the competition as yeast proteins in the provided database (see Table 1).
| Samples
| |||||||
| Name | Origin | Molecular Weight | 1 | 2 | 3 | 4 | |
| A | Ovalbumin | Egg White | 45 KD | 65 | 55 | 15 | 2 |
| B | Myoglobin | Equine Heart | 17 KD | 55 | 15 | 2 | 65 |
| C | Phosphorylase b | Rabbit Muscle | 97 KD | 15 | 2 | 65 | 55 |
| D | Beta-Glactosidase | Escherichia Coli | 116 KD | 2 | 65 | 55 | 15 |
| E | Bovine Serum Albumin | Bovine Serum | 66 KD | 11 | 0.6 | 10 | 500 |
| F | Carbonic Anhydrase | Bovine Erythrocytes | 29 KD | 10 | 500 | 11 | 0.6 |
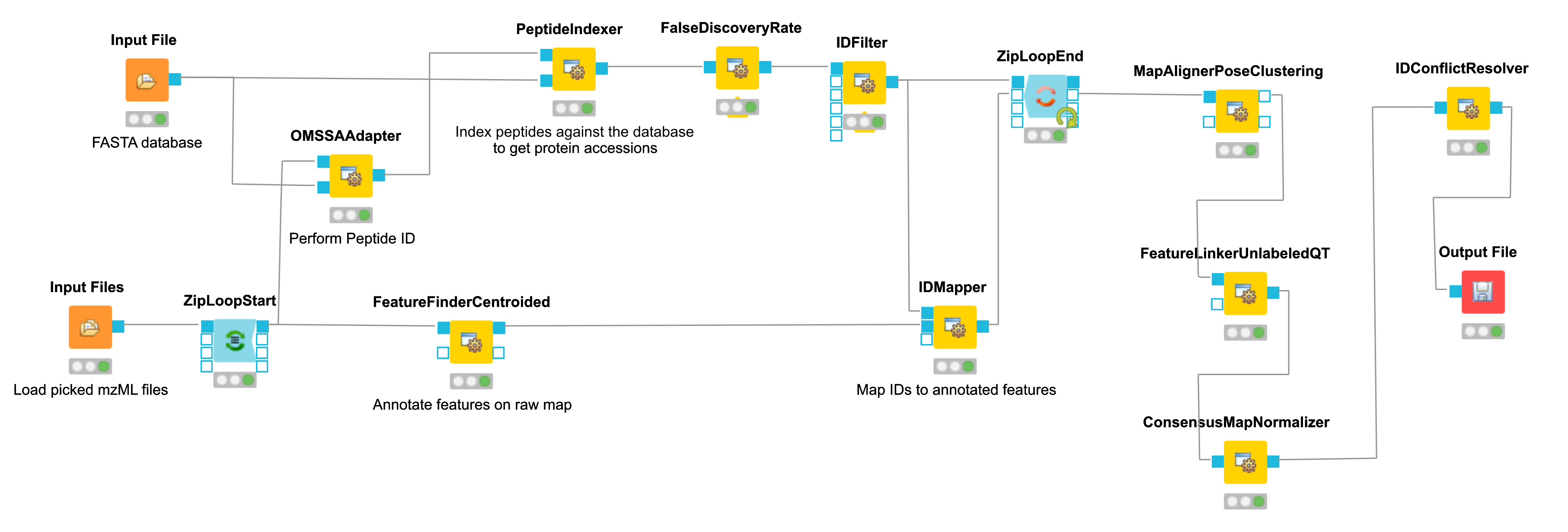
The iPRG LFQ workflow (Fig. 18) consists of an identification and a quantification part. The
identification is achieved by searching the computationally calculated MS2 spectra from
a sequence database (Input File node, here with the given database from iPRG:
 )
against the MS2 from the original data (Input Files node with all mzMLs following
)
against the MS2 from the original data (Input Files node with all mzMLs following
 )
using the OMSSAAdapter.
)
using the OMSSAAdapter.
Note: If you want to reproduce the results at home, you have to download the iPRG data in mzML format and perform Peakpicking on it. Or convert and pick the raw data with msconvert.
Afterwards the results are scored using the FalseDiscoveryRate node and filtered to obtain only unique peptides (IDFilter) since MSstats does not support shared peptides, yet. The quantification is achieved by the FeatureFinderCentroided, which performs the feature detection on the samples (maps). In the end the quantification results are combined with the filtered identification results (IDMapper). In addition, a linear retention time alignment is performed (MapAlignerPoseClustering), followed by the feature linking process (FeatureLinkerUnlabledQT). The ConsensusMapNormalizer is used to normalize the intensities via robust regression over a set of maps and the IDConflictResolver assures that only one identification (best score) is associated with a feature. The output of this workflow is a consensusXML file, which can now be converted using the MSstatsConverter (see section 3.5.5).
As mentioned before, the downstream analysis can be performed using MSstats. In this case an experimental design has to be specified for the OpenMS workflow. The structure of the experimental design used in OpenMS in case of the iPRG dataset is specified in Table 2. An explanation of the variables can be found in Table 3.
| Fraction_Group | Fraction | Spectra_Filepath | Label | Sample |
| 1 | 1 | Sample1-A | 1 | 1 |
| 2 | 1 | Sample1-B | 1 | 2 |
| 3 | 1 | Sample1-C | 1 | 3 |
| 4 | 1 | Sample2-A | 1 | 4 |
| 5 | 1 | Sample2-B | 1 | 5 |
| 6 | 1 | Sample2-C | 1 | 6 |
| 7 | 1 | Sample3-A | 1 | 7 |
| 8 | 1 | Sample3-B | 1 | 8 |
| 9 | 1 | Sample3-C | 1 | 9 |
| 10 | 1 | Sample4-A | 1 | 10 |
| 11 | 1 | Sample4-B | 1 | 11 |
| 12 | 1 | Sample4-C | 1 | 12 |
| Sample | MSstats_Condition | MSstats_BioReplicate | ||
| 1 | 1 | 1 | ||
| 2 | 1 | 2 | ||
| 3 | 1 | 3 | ||
| 4 | 2 | 4 | ||
| 5 | 2 | 5 | ||
| 6 | 2 | 6 | ||
| 7 | 3 | 7 | ||
| 8 | 3 | 8 | ||
| 9 | 3 | 9 | ||
| 10 | 4 | 10 | ||
| 11 | 4 | 11 | ||
| 12 | 4 | 12 |
| variables | value |
| Fraction_Group | Index used to group fractions and source files. |
| Fraction | 1st, 2nd, .., fraction. Note: All runs must have the same number of fractions. |
| Spectra_Filepath | Path to mzML files |
| Label | label-free: always 1 |
| TMT6Plex: 1...6 |
|
| SILAC with light and heavy: 1..2 |
|
| Sample | Index of sample measured in the specified label X, in fraction Y of fraction group Z. |
| Conditions | Further specification of different conditions (e.g. MSstats_Condition; MSstats_BioReplicate) |
The conditions are highly dependent on the type of experiment and on which kind of analysis you want to perform. For the MSstats analysis the information which sample belongs to which condition and if there are biological replicates are mandatory. This can be specified in further condition columns as explained in Table 3. For a detailed description of the MSstats-specific terminology, see their documentation e.g. in the R vignette.
Conversion of the OpenMS-internal consensusXML format (which is an aggregation of quantified and possibly identified features across several MS-maps) to a table (in MSstats-conformant CSV format) is very easy. First, create a new KNIME workflow. Then, run the MSstatsConverter node with a consensusXML and the manually created (e.g. in Excel) experimental design as inputs (loaded via Input File nodes). The first input can be found in

This file was generated by using the  workflow (seen in Fig. 18). The second input is specified in
workflow (seen in Fig. 18). The second input is specified in
 .
.
Adjust the parameters in the config dialog of the converter to match the given experimental design file and to use a simple summing for peptides that elute in multiple features (with the same charge state, i.e. m/z value).
| parameter | value |
| msstats_bioreplicate | MSstats_Bioreplicate |
| msstats_condition | MSstats_Condition |
| labeled_reference_peptides | false |
| retention_time_summarization_method (advanced) | sum |
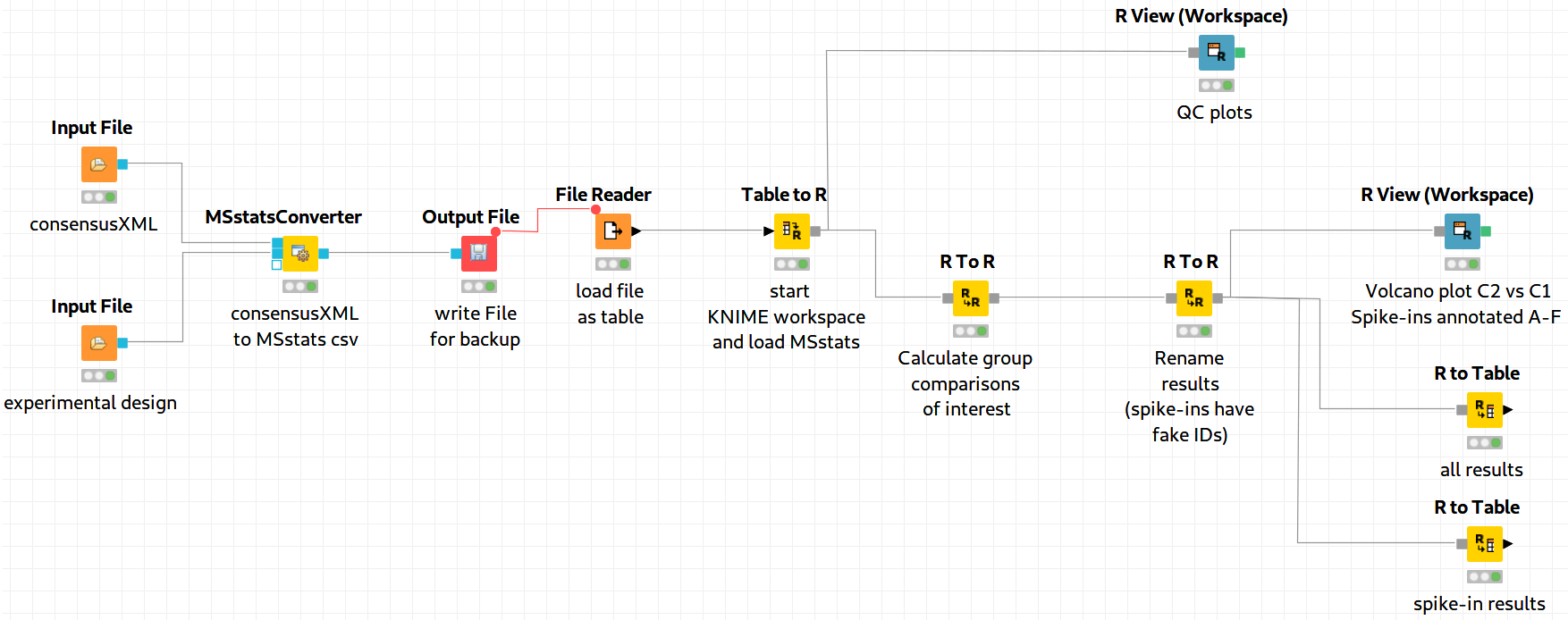
The downstream analysis of the peptide ions with MSstats is performed in several steps. These steps are reflected by several KNIME R nodes, which consume the output of MSstatsConverter. The outline of the workflow is shown in Figure 19.
We load the file resulting from MSStatsConverter either by saving it with an Output File node and reloading it with the File Reader. Or for advanced users, you can use a URI Port to Variable node and use the variable in the File Reader config dialog (V button - located on the right of the ”Browse...” button) to read from the temporary file.
Preprocessing
The first node (Table to R) loads MSstats as well as the data from the previous KNIME node
and performs a preprocessing step on the input data. The inline R script (that needs to be
pasted into the config dialog of the node)
allows further preparation of the data produced by MSstatsConverter before the
actual analysis is performed. In this example, the lines with proteins, which were
identified with only one feature, were retained. Alternatively they could be removed.
In the same node, most importantly, the following line:
transforms the data into a format that is understood by MSstats. Here, dataProcess is one of the most important functions that the R package provides. The function performs the following steps:
In this example, we just state that missing intensity values are represented by the ’NA’ string.
Group Comparison
The goal of the analysis is the determination of differentially-expressed proteins among the
different conditions C1-C4. We can specify the comparisons that we want to make in a
comparison matrix. For this, let’s consider the following example:
 | (3.1) |
This matrix has the following properties:
 .
.We can generate such a matrix in R using the following code snippet in (for example) a new R to R node that takes over the R workspace from the previous node with all its variables:
comparison1<-matrix(c(-1,1,0,0),nrow=1) comparison2<-matrix(c(-1,0,1,0),nrow=1) comparison3<-matrix(c(-1,0,0,1),nrow=1) comparison4<-matrix(c(0,-1,1,0),nrow=1) comparison5<-matrix(c(0,-1,0,1),nrow=1) comparison6<-matrix(c(0,0,-1,1),nrow=1) comparison <- rbind(comparison1, comparison2, comparison3, comparison4, comparison5, comparison6) row.names(comparison)<-c("C2-C1","C3-C1","C4-C1","C3-C2","C4-C2","C4-C3")
Here, we assemble each row in turn, concatenate them at the end, and provide row names for labeling the rows with the respective condition.
In MSstats, the group comparison is then performed with the following line:
No more parameters need to be set for performing the comparison.
Result Processing
In a next R to R node, the results are being processed. The following code snippet:
test.MSstats.cr <- test.MSstats$ComparisonResult # Rename spiked ins to A,B,C.... pnames <- c("A", "B", "C", "D", "E", "F") names(pnames) <- c( "sp|P44015|VAC2_YEAST", "sp|P55752|ISCB_YEAST", "sp|P44374|SFG2_YEAST", "sp|P44983|UTR6_YEAST", "sp|P44683|PGA4_YEAST", "sp|P55249|ZRT4_YEAST" ) test.MSstats.cr.spikedins <- bind_rows( test.MSstats.cr[grep("P44015", test.MSstats.cr$Protein),], test.MSstats.cr[grep("P55752", test.MSstats.cr$Protein),], test.MSstats.cr[grep("P44374", test.MSstats.cr$Protein),], test.MSstats.cr[grep("P44683", test.MSstats.cr$Protein),], test.MSstats.cr[grep("P44983", test.MSstats.cr$Protein),], test.MSstats.cr[grep("P55249", test.MSstats.cr$Protein),] ) # Rename Proteins test.MSstats.cr.spikedins$Protein <- sapply(test.MSstats.cr.spikedins$Protein, function(x) {pnames[as.character(x)]}) test.MSstats.cr$Protein <- sapply(test.MSstats.cr$Protein, function(x) { x <- as.character(x) if (x %in% names(pnames)) { return(pnames[as.character(x)]) } else { return("") } })
will rename the spiked-in proteins to A,B,C,D,E, and F and remove the names of other proteins, which will be beneficial for the subsequent visualization, as for example performed in Figure 20.
Export
The last four nodes, each connected and making use of the same workspace from
the last node, will export the results to a textual representation and volcano plots
for further inspection. Firstly, quality control can be performed with the following
snippet:
The code for this snippet is embedded in the first output node of the workflow. The resulting boxplots show the log2 intensity distribution across the MS runs.
The second node is an R View (Workspace) node that returns a Volcano plot which displays differentially expressed proteins between conditions C2 vs. C1. The plot is described in more detail in the following Result section. This is how you generate it:
The last two nodes export the MSstats results as a KNIME table for potential further analysis or for writing it to a (e.g. csv) file. Note that you could also write output inside the Rscript if you are familiar with it. Use the following for an R to Table node exporting all results:
And this for an R to Table node exporting only results for the spike-ins:
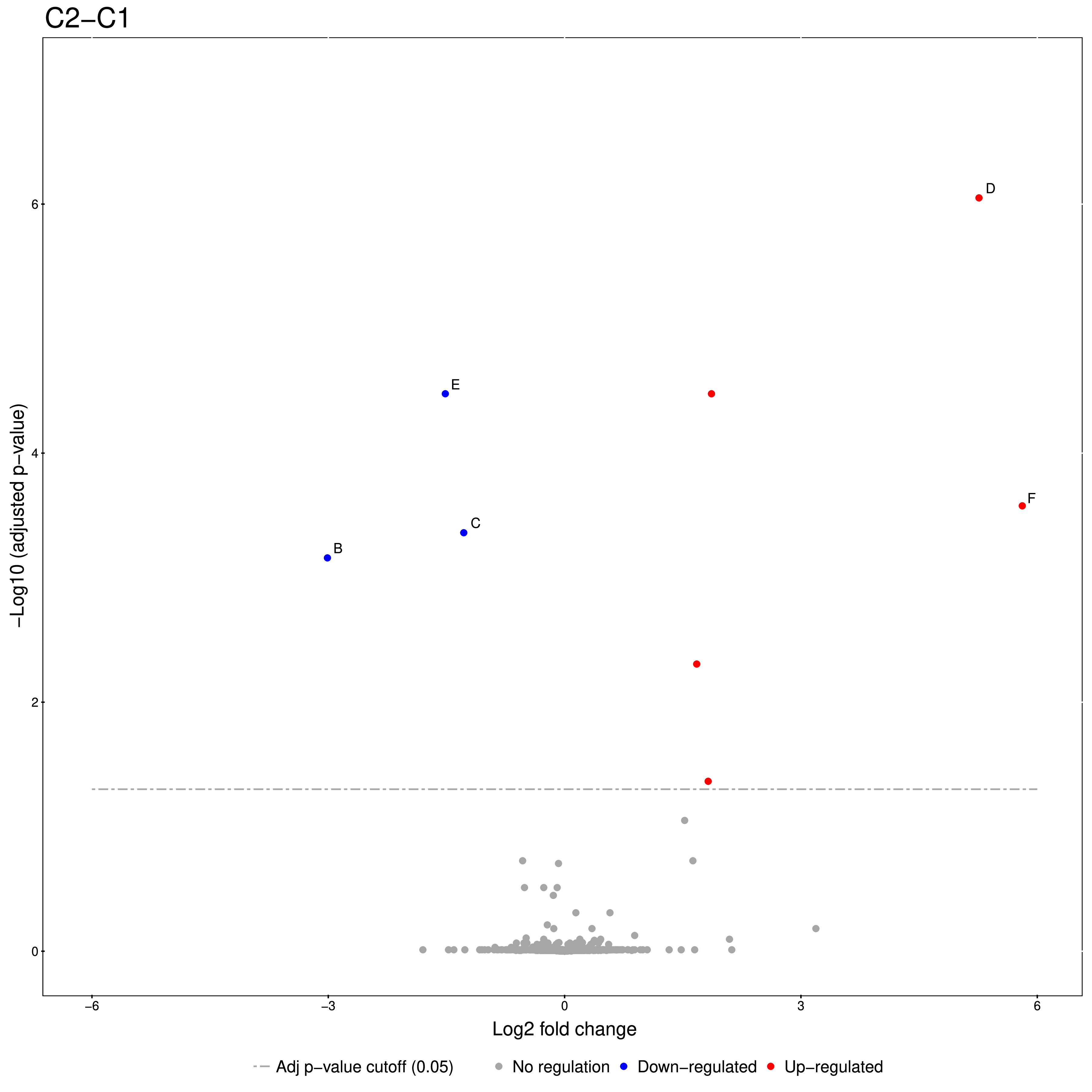
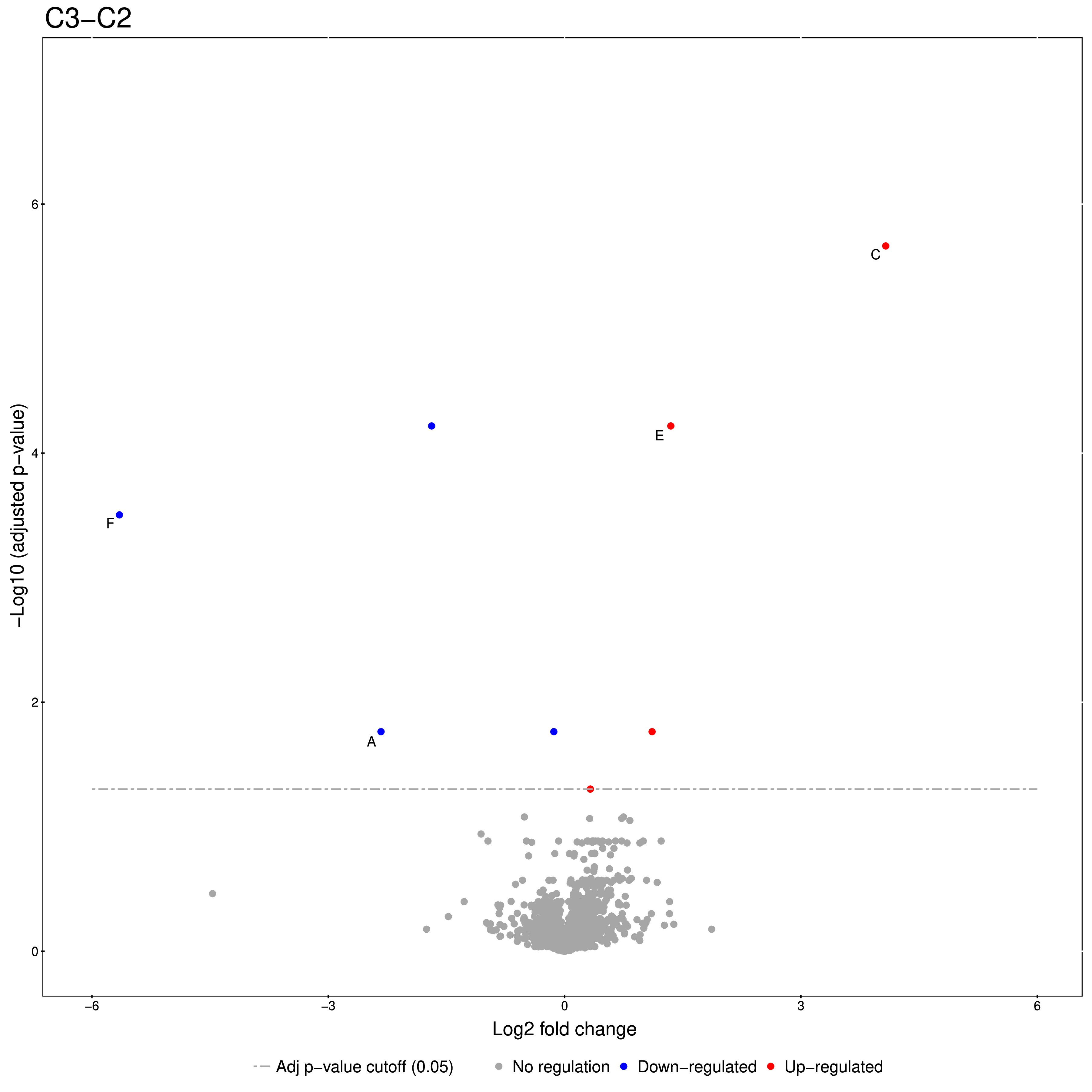
An excerpt of the main result of the group comparison can be seen in Figure 20.
The Volcano plots show differently expressed spiked-in proteins. In the left plot, which shows
the fold-change C2-C1, we can see the proteins D and F (sp|P44983|UTR6_YEAST and
sp|P55249|ZRT4_YEAST) are significantly over-expressed in C2, while the proteins B,C, and E
(sp|P55752|ISCB_YEAST, sp|P55752|ISCB_YEAST, and sp|P44683|PGA4_YEAST) are
under-expressed. In the right plot, which shows the fold-change ratio of C3 vs. C2, we can see
the proteins E and C (sp|P44683|PGA4_YEAST and sp|P44374|SFG2_YEAST) over-expressed and
the proteins A and F (sp|P44015|VAC2_YEAST and sp|P55249|ZRT4_YEAST) under-expressed.
The plots also show further differentially-expressed proteins, which do not belong to the
spiked-in proteins.
The full analysis workflow can be found under .
.
In the last chapter, we have successfully quantified peptides in a label-free experiment. As a
next step, we will further extend this label-free quantification workflow by protein inference and
protein quantification capabilities. This workflow uses some of the more advanced concepts of
KNIME, as well as a few more nodes containing R code. For these reasons, you will not have to
build it yourself. Instead, we have already prepared and copied this workflow to the USB sticks.
Just import  into
KNIME via the menu entry
into
KNIME via the menu entry  and
double-click the imported workflow in order to open it.
and
double-click the imported workflow in order to open it.
Before you can execute the workflow, you again have to correct the locations of the files in the Input Files nodes (don’t forget the one for the FASTA database inside the “ID” meta node). Try and run your workflow by executing all nodes at once.
We have made the following changes compared to the original label-free quantification workflow from the last chapter:
 .
.
 ). However, the meta node contains an additional subworkflow which, besides calling
FidoAdapter, performs a statistical validation (e.g. (pseudo) receiver operating
curves; ROCs) of the protein inference results using some of the more advanced
KNIME and R nodes. The meta node also shows how to use MzTabExporter and
MzTabReader.
). However, the meta node contains an additional subworkflow which, besides calling
FidoAdapter, performs a statistical validation (e.g. (pseudo) receiver operating
curves; ROCs) of the protein inference results using some of the more advanced
KNIME and R nodes. The meta node also shows how to use MzTabExporter and
MzTabReader.
In the following, we will explain the subworkflow contained in the Protein inference with FidoAdapter meta node.
For downstream analysis on the protein ID level in KNIME, it is again necessary to convert the idXML-file-format result generated from FidoAdapter into a KNIME table.
ROC Curves (Receiver Operating Characteristic curves) are graphical plots that
visualize sensitivity (true-positive rate) against fall-out (false positive rate). They
are often used to judge the quality of a discrimination method like e.g., peptide or
protein identification engines. ROC Curve already provides the functionality of drawing
ROC curves for binary classification problems. When configuring this node, select the
opt_global_target_decoy column as the class (i.e. target outcome) column. We want to find out,
how good our inferred protein probability discriminates between them, therefore
add
best_search_engine_score[1] (the inference engine score is treated like a peptide search engine
score) to the list of ”Columns containing positive class probabilities”. View the plot by
right-clicking and selecting  . A
perfect classifier has an area under the curve (AUC) of 1.0 and its curve touches the upper left
of the plot. However, in protein or peptide identification, the ground-truth (i.e., which target
identifications are true, which are false) is usually not known. Instead, so called pseudo-ROC
Curves are regularly used to plot the number of target proteins against the false discovery rate
(FDR) or its protein-centric counterpart, the q-value. The FDR is approximated by using the
target-decoy estimate in order to distinguish true IDs from false IDs by separating target IDs
from decoy IDs.
. A
perfect classifier has an area under the curve (AUC) of 1.0 and its curve touches the upper left
of the plot. However, in protein or peptide identification, the ground-truth (i.e., which target
identifications are true, which are false) is usually not known. Instead, so called pseudo-ROC
Curves are regularly used to plot the number of target proteins against the false discovery rate
(FDR) or its protein-centric counterpart, the q-value. The FDR is approximated by using the
target-decoy estimate in order to distinguish true IDs from false IDs by separating target IDs
from decoy IDs.
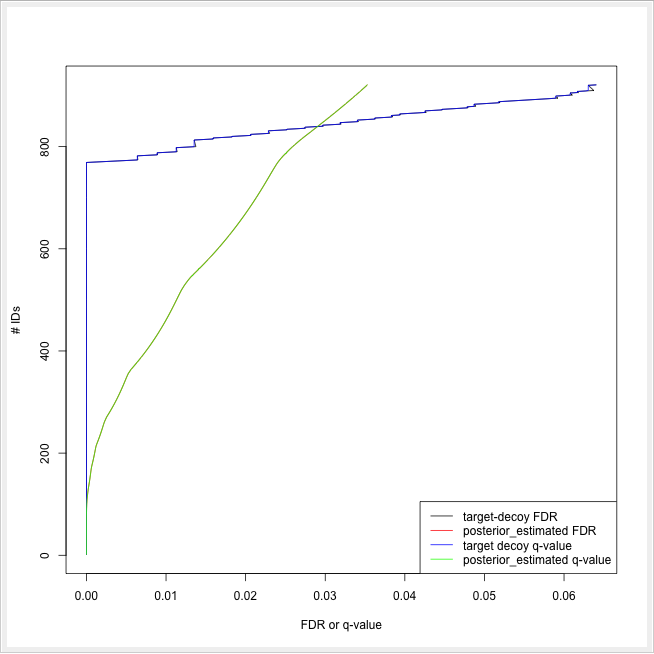
ROC curves illustrate the discriminative capability of the scores of IDs. In the case of protein identifications, Fido produces the posterior probability of each protein as the output score. However, a perfect score should not only be highly discriminative (distinguishing true from false IDs), it should also be “calibrated” (for probability indicating that all IDs with reported posterior probability scores of 95% should roughly have a 5% probability of being false. This implies that the estimated number of false positives can be computed as the sum of posterior error probabilities ( = 1 - posterior probability) in a set, divided by the number of proteins in the set. Thereby a posterior-probability-estimated FDR is computed which can be compared to the actual target-decoy FDR. We can plot calibration curves to help us visualize the quality of the score (when the score is interpreted as a probability as Fido does), by comparing how similar the target-decoy estimated FDR and the posterior probability estimated FDR are. Good results should show a close correspondence between these two measurements, although a non-correspondence does not necessarily indicate wrong results.
The calculation is done by using a simple R script in R snippet. First, the target decoy protein FDR is computed as the proportion of decoy proteins among all significant protein IDs. Then posterior probabilistic-driven FDR is estimated by the average of the posterior error probability of all significant protein IDs. Since FDR is the property for a group of protein IDs, we can also calculate a local property for each protein: the q-value of a certain protein ID is the minimum FDR of any groups of protein IDs that contain this protein ID. We plot the protein ID results versus two different kinds of FDR estimates in R View(Table) (see Fig. 22).
In the last chapters, we identified and quantified peptides in a label-free experiment. In this section, we would like to introduce a possible workflow for the analysis of isobaric data.
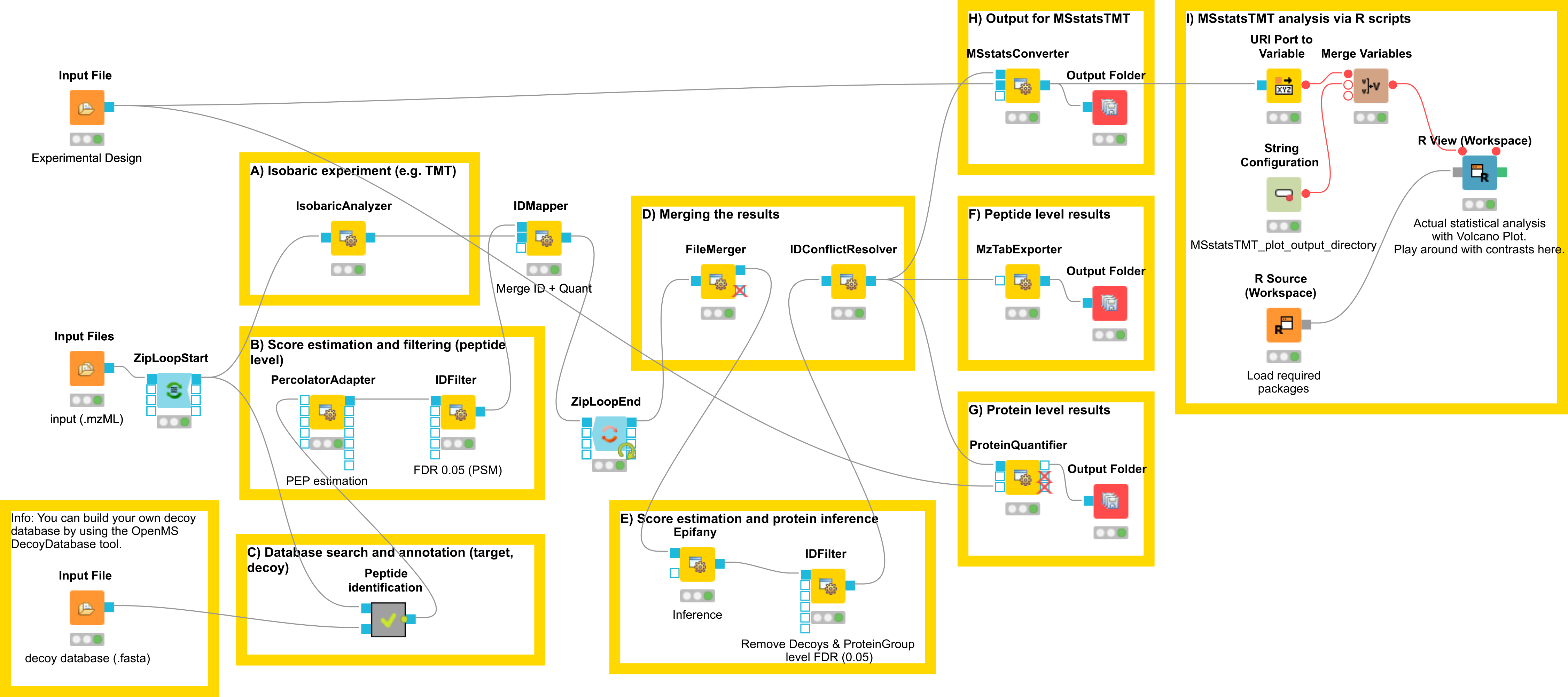
Let’s have a look at the workflow (see Fig 23)
The full analysis workflow can be found here:  .
.
The workflow has four input nodes. The first for the experimental design to allow for MSstatsTMT compatible export (MSstatsConverter). The second for the .mzML files with the centroided spectra from the isobaric labeling experiment and the third one for the .fasta database used for identification. The last one allows to specify an output path for the plots generated by the R View, which runs MSstatsTMT (I). The quantification (A) is performed using the IsobaricAnalzyer. The tool is able to extract and normalize quantitative information from TMT and iTRAQ data. The values can be assessed from centroided MS2 or MS3 spectra (if available). Isotope correction is performed based on the specified correction matrix (as provided by the manufacturer). The identification (C) is applied as known from the previous chapters by using database search and a target-decoy database.
To reduce the complexity of the data for later inference the q-value estimation and FDR
filtering is performed on PSM level for each file individually (B). Afterwards the identification
(PSM) and quantiative information is combined using the IDMapper. After the processing of all
available files, the intermediate results are aggregated (FileMerger - D). All PSM results are
used for score estimation and protein inference (Epifany) (E). For detailed information about
protein inference please see Chaper 4. Then, decoys are removed and the inference
results are filtered via a protein group FDR. Peptide level results can be exported via
MzTabExporter (F), protein level results can be obtained via the ProteinQuantifier (G)
or the results can exported (MSstatsConverter - H) and further processed with
the following R pipeline to allow for downstream processing using MSstatsTMT.
Please import the workflow from  into
KNIME via the menu entry
into
KNIME via the menu entry  and
double-click the imported workflow in order to open it. Before you can execute the workflow,
you have to correct the locations of the files in the Input Files nodes (don’t forget the one for
the FASTA database inside the “ID” meta node). Try and run your workflow by executing all
nodes at once.
and
double-click the imported workflow in order to open it. Before you can execute the workflow,
you have to correct the locations of the files in the Input Files nodes (don’t forget the one for
the FASTA database inside the “ID” meta node). Try and run your workflow by executing all
nodes at once.
The R package MSstatsTMT can be used for protein significance analysis in shotgun mass
spectrometry-based proteomic experiments with tandem mass tag (TMT) labeling.
MSstatsTMT provides functionality for two types of analysis & their visualization: Protein
summarization based on peptide quantification and Model-based group comparison to
detect significant changes in abundance. It depends on accurate feature detection,
identification and quantification which can be performed e.g. by an OpenMS workflow.
In general MSstatsTMT can be used for data processing & visualization, as well as statistical
modeling. Please see [13] and http://msstats.org/msstatstmt/ for further information.
There is also a very helpful online lecture and tutorial for MSstatsTMT from the May Institute Workshop 2020. Please see https://youtu.be/3CDnrQxGLbA
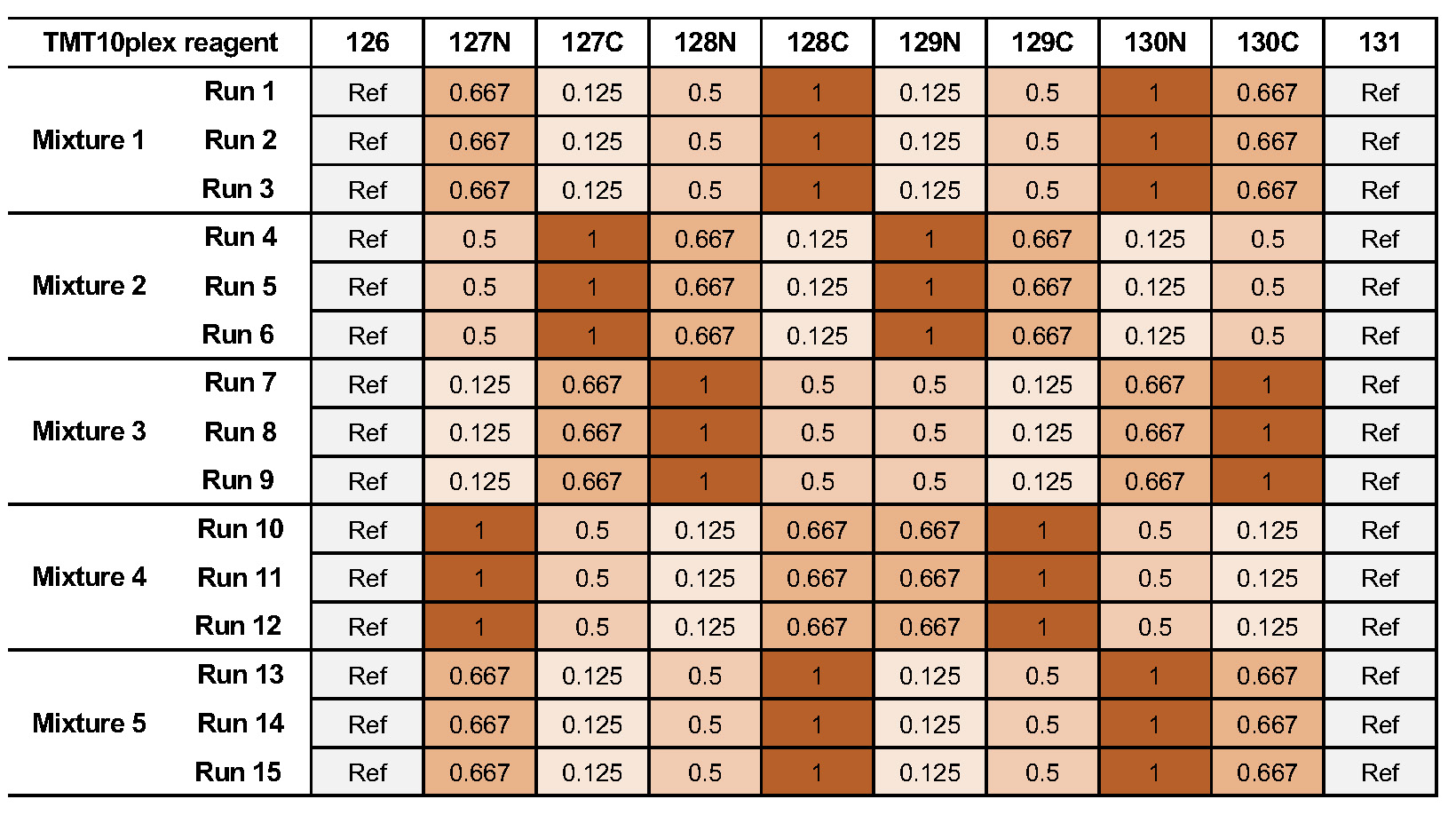
We are using the MSV000084264 ground truth dataset, which consits of TMT10plex controlled mixes of different concentrated UPS1 peptides spiked into SILAC HeLa peptides measured in a dilution series https://www.omicsdi.org/dataset/massive/MSV000084264. Figure 24 shows the experimental design. In this experiment 5 different TMT10plex mixtures – different labeling strategies – were analysed. These were measured in triplicates represented by the 15 MS runs (3 runs each). The example data, database and experimental design to run the workflow can be found here https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Tutorials/Data/isobaric_MSV000084264/.
The experimental design in table format allows for MSstatsTMT compatible export. The design is represented by two tables. The first one 4 represents the overall structure of the experiment in terms of samples, fractions, labels and fraction groups. The second one 5 adds to the first by specifying specific conditions, biological replicates as well as mixtures and label for each channel. For additional information about the experimental design please see Table 3 in Chapter 3.5.4.
| Spectra_Filepath | Fraction | Label | Fraction_Group | Sample |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 1 | 1 | 1 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 2 | 1 | 2 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 3 | 1 | 3 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 4 | 1 | 4 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 5 | 1 | 5 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 6 | 1 | 6 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 7 | 1 | 7 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 8 | 1 | 8 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 9 | 1 | 9 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_01.mzML | 1 | 10 | 1 | 10 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 1 | 2 | 11 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 2 | 2 | 12 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 3 | 2 | 13 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 4 | 2 | 14 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 5 | 2 | 15 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 6 | 2 | 16 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 7 | 2 | 17 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 8 | 2 | 18 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 9 | 2 | 19 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_02.mzML | 1 | 10 | 2 | 20 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 1 | 3 | 21 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 2 | 3 | 22 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 3 | 3 | 23 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 4 | 3 | 24 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 5 | 3 | 25 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 6 | 3 | 26 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 7 | 3 | 27 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 8 | 3 | 28 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 9 | 3 | 29 |
| 161117_SILAC_HeLa_UPS1_TMT10_SPS_MS3_Mixture1_03.mzML | 1 | 10 | 3 | 30 |
| Sample | MSstats_Condition | MSstats_BioReplicate | MSstats_Mixture | LabelName |
| 1 | Norm | Norm | 1 | 126 |
| 2 | 0.667 | 0.667 | 1 | 127N |
| 3 | 0.125 | 0.125 | 1 | 127C |
| 4 | 0.5 | 0.5 | 1 | 128N |
| 5 | 1 | 1 | 1 | 128C |
| 6 | 0.125 | 0.125 | 1 | 129N |
| 7 | 0.5 | 0.5 | 1 | 129C |
| 8 | 1 | 1 | 1 | 130N |
| 9 | 0.667 | 0.667 | 1 | 130C |
| 10 | Norm | Norm | 1 | 131 |
| 11 | Norm | Norm | 1 | 126 |
| 12 | 0.667 | 0.667 | 1 | 127N |
| 13 | 0.125 | 0.125 | 1 | 127C |
| 14 | 0.5 | 0.5 | 1 | 128N |
| 15 | 1 | 1 | 1 | 128C |
| 16 | 0.125 | 0.125 | 1 | 129N |
| 17 | 0.5 | 0.5 | 1 | 129C |
| 18 | 1 | 1 | 1 | 130N |
| 19 | 0.667 | 0.667 | 1 | 130C |
| 20 | Norm | Norm | 1 | 131 |
| 21 | Norm | Norm | 1 | 126 |
| 22 | 0.667 | 0.667 | 1 | 127N |
| 23 | 0.125 | 0.125 | 1 | 127C |
| 24 | 0.5 | 0.5 | 1 | 128N |
| 25 | 1 | 1 | 1 | 128C |
| 26 | 0.125 | 0.125 | 1 | 129N |
| 27 | 0.5 | 0.5 | 1 | 129C |
| 28 | 1 | 1 | 1 | 130N |
| 29 | 0.667 | 0.667 | 1 | 130C |
| 30 | Norm | Norm | 1 | 131 |
After running the worklfow the MSstatsConverter will convert the OpenMS output in addition with the experimental design to a file (.csv) which can be processed by using MSstatsTMT.
Here, we depict the analysis by MSstatsTMT using a segment of the isobaric analysis workflow
(Fig. 25 ). The segment is available as  .
.
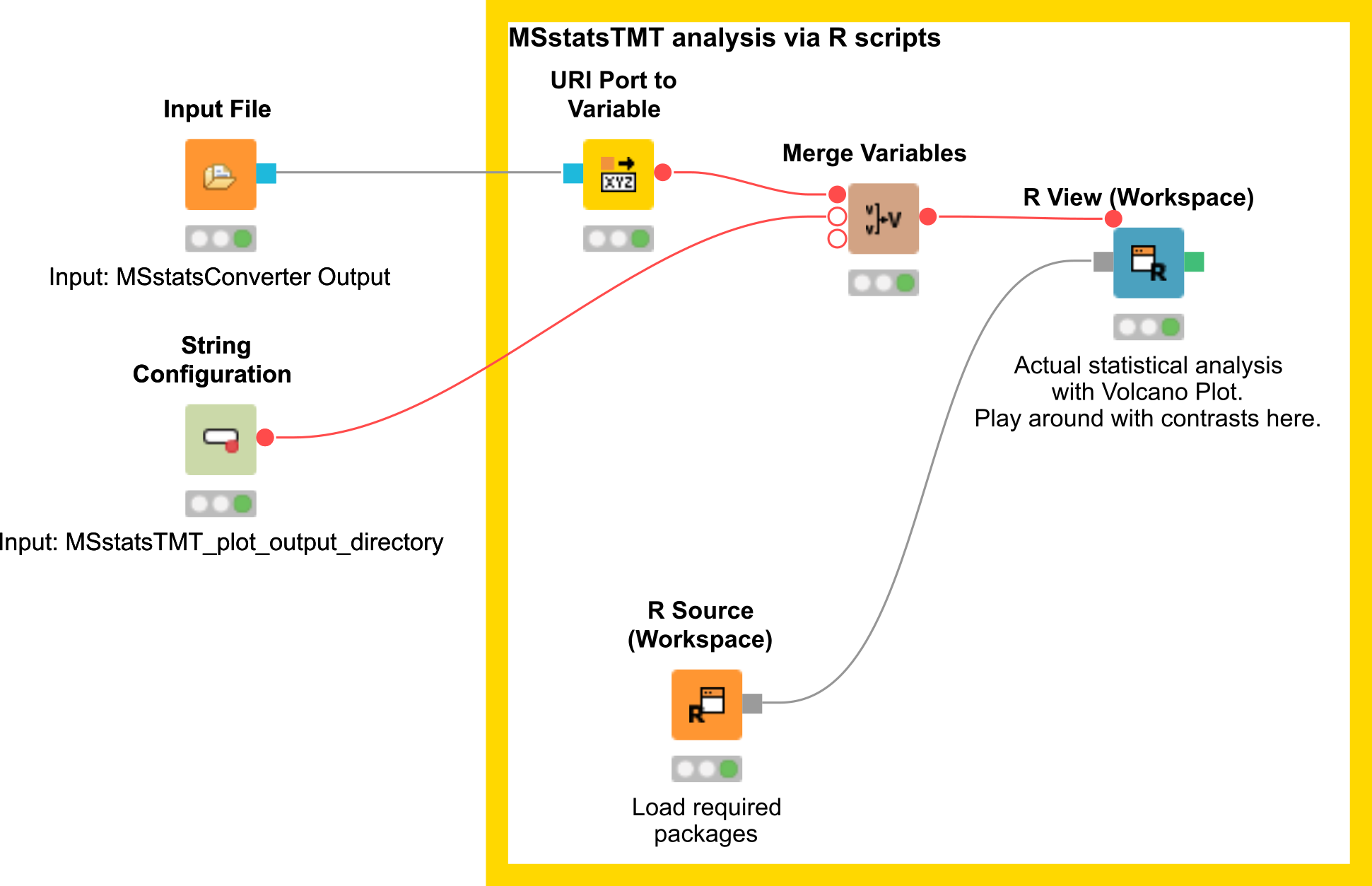
There are two input nodes, the first one takes the result (.csv) from the MSstatsConverter and the second a path to the directory where the plots generated by MSstatsTMT should be saved. The R source node loads the required packages, such as dplyr for data wrangling, MSstatsTMT for analysis and MSstats for plotting. The inputs are further processed in the R View node.
Here, the data of the Input File is loaded into R using the flow variable [”URI-0”]:
The OpenMStoMSstatsTMTFormat function preprocesses the OpenMS report and converts it into the required input format for MSstatsTMT, by filtering based on unique peptides and measurments in each MS run.
Afterwards different normalization steps are performed (global, protein, runs) as well as data imputation by using the msstats method. In addition peptide level data is summarized to protein level data.
There a lot of different possibilities to configure this method please have a look at the MSstatsTMT package for additional detailed information http://bioconductor.org/packages/release/bioc/html/MSstatsTMT.html
The next step is the comparions of the different conditions, here either a pairwise comparision can be performed or a confusion matrix can be created. The goal is to detect and compare the UPS peptides spiked in at different concentrations.
# prepare contrast matrix unique(quant.data$Condition) comparison<-matrix(c(-1,0,0,1, 0,-1,0,1, 0,0,-1,1, 0,1,-1,0, 1,-1,0,0), nrow=5, byrow = T) # Set the names of each row row.names(comparison)<- contrasts <- c("1-0125", "1-05", "1-0667", "05-0667", "0125-05") # Set the column names colnames(comparison)<- c("0.125", "0.5", "0.667", "1")
The constructed confusion matrix is used in the groupComparisonTMT function to test for significant changes in protein abundance across conditions based on a family of linear mixed-effects models in TMT experiments.
In the next step the comparison can be plotted using the groupComparisonPlots function by MSstats
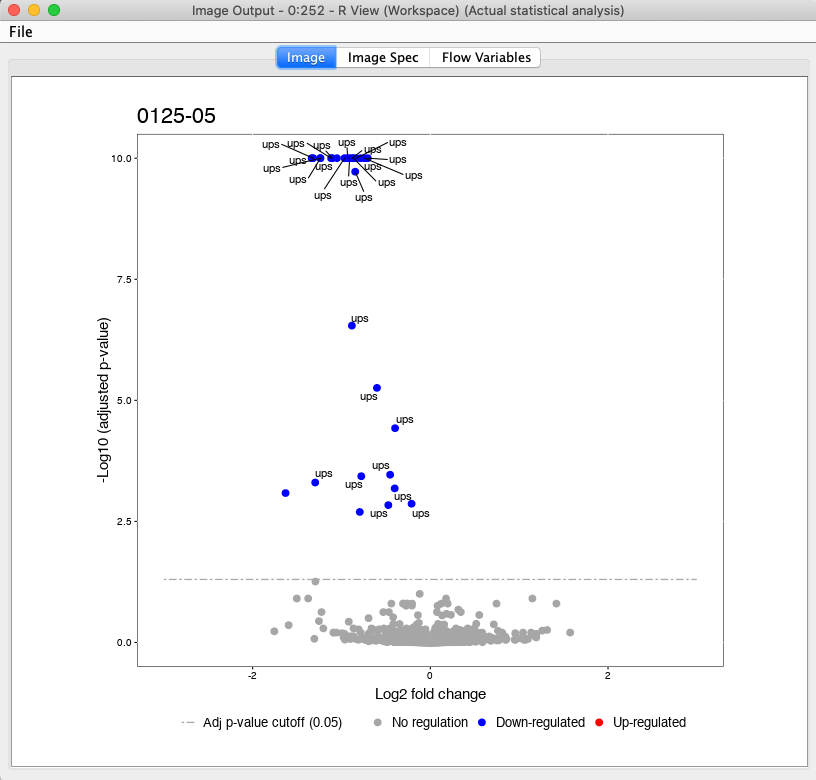
Here, we have a example output of the R View, which depicts the significant regulated UPS proteins in the comparison of 125 to 05 (Fig. 26).
All plots are saved to the in the beginning specified output directory in addition.
The isobaric analysis does not always has to be performed on protein level, for example for
phosphoproteomics studies one is usually interested on the peptide level - in addition inference
on peptides with post-translational modification is not straight forward. Here, we present and
additonal workflow on peptide level, which can potentially be adapted and used for such cases.
Please see  .
.
Quantification and identification of chemical compounds are basic tasks in metabolomic studies. In this tutorial session we construct a UPLC-MS based, label-free quantification and identification workflow. Following quantification and identification we then perform statistical downstream analysis to detect quantification values that differ significantly between two conditions. This approach can, for example, be used to detect biomarkers. Here, we use two spike-in conditions of a dilution series (0.5 mg/l and 10.0 mg/l, male blood background, measured in triplicates) comprising seven isotopically labeled compounds. The goal of this tutorial is to detect and quantify these differential spike-in compounds against the complex background.
For the metabolite quantification we choose an approach similar to the one used for peptides, but this time based on the OpenMS FeatureFinderMetabo method. This feature finder again collects peak picked data into individual mass traces. The reason why we need a different feature finder for metabolites lies in the step after trace detection: the aggregation of isotopic traces belonging to the same compound ion into the same feature. Compared to peptides with their averagine model, small molecules have very different isotopic distributions. To group small molecule mass traces correctly, an aggregation model tailored to small molecules is thus needed.
 .
.
 and connect the first output port of the Input File to the FeatureFinderMetabo.
and connect the first output port of the Input File to the FeatureFinderMetabo.
In the following advanced parameters will be highlighted. These parameter can be altered
if the  field in the specific tool is activated (right bottom corner - see 2.4.2).
field in the specific tool is activated (right bottom corner - see 2.4.2).
| parameter | value |
| algorithm →common →chrom_fwhm | 8.0 |
| algorithm →mtd →trace_termination_criterion | sample_rate |
| algorithm →mtd →min_trace_length | 3.0 |
| algorithm →mtd →max_trace_length | 600.0 |
| algorithm →epd →width_filtering | off |
| algorithm →ffm →report_convex_hulls | true |
The parameters change the behavior of FeatureFinderMetabo as follows:
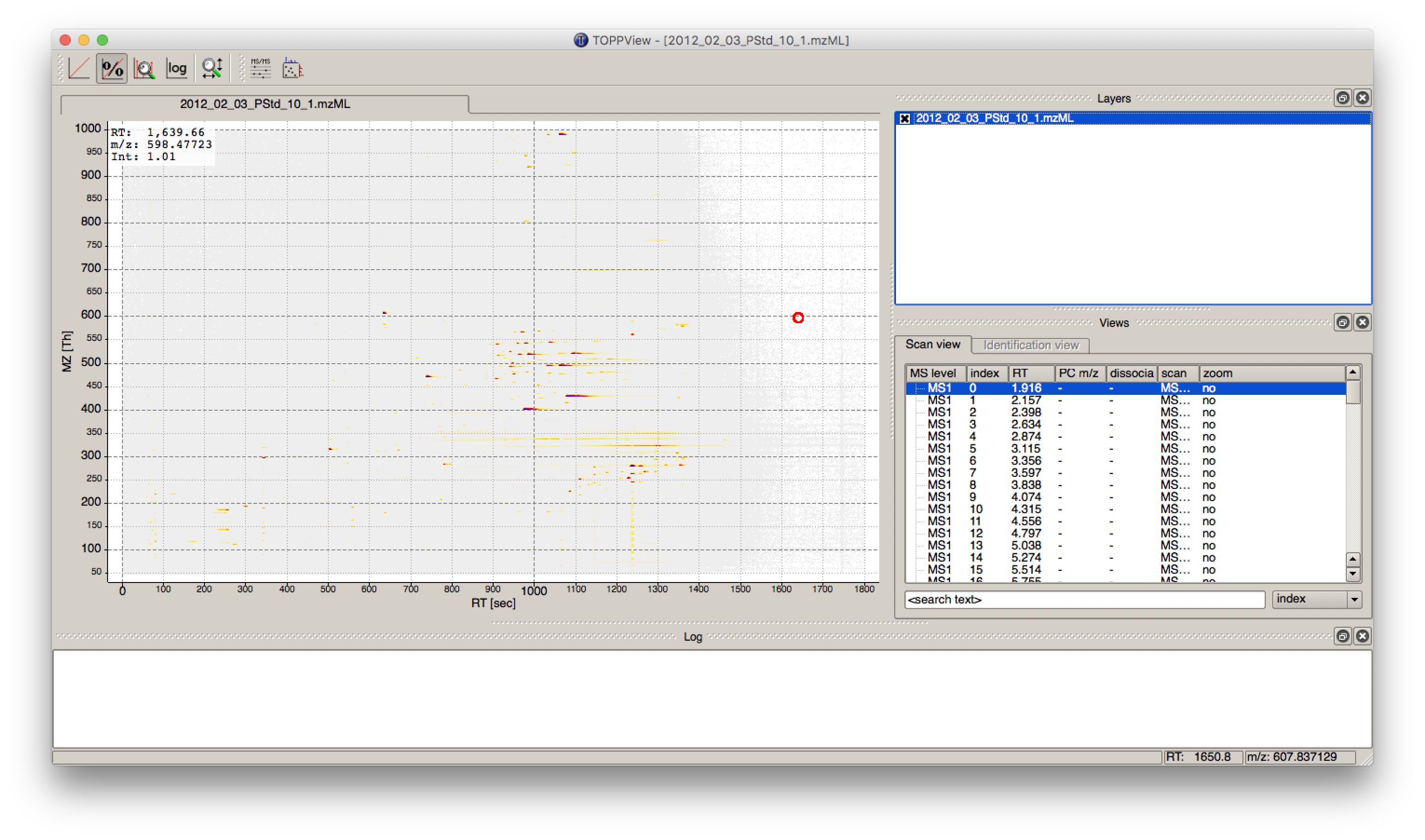
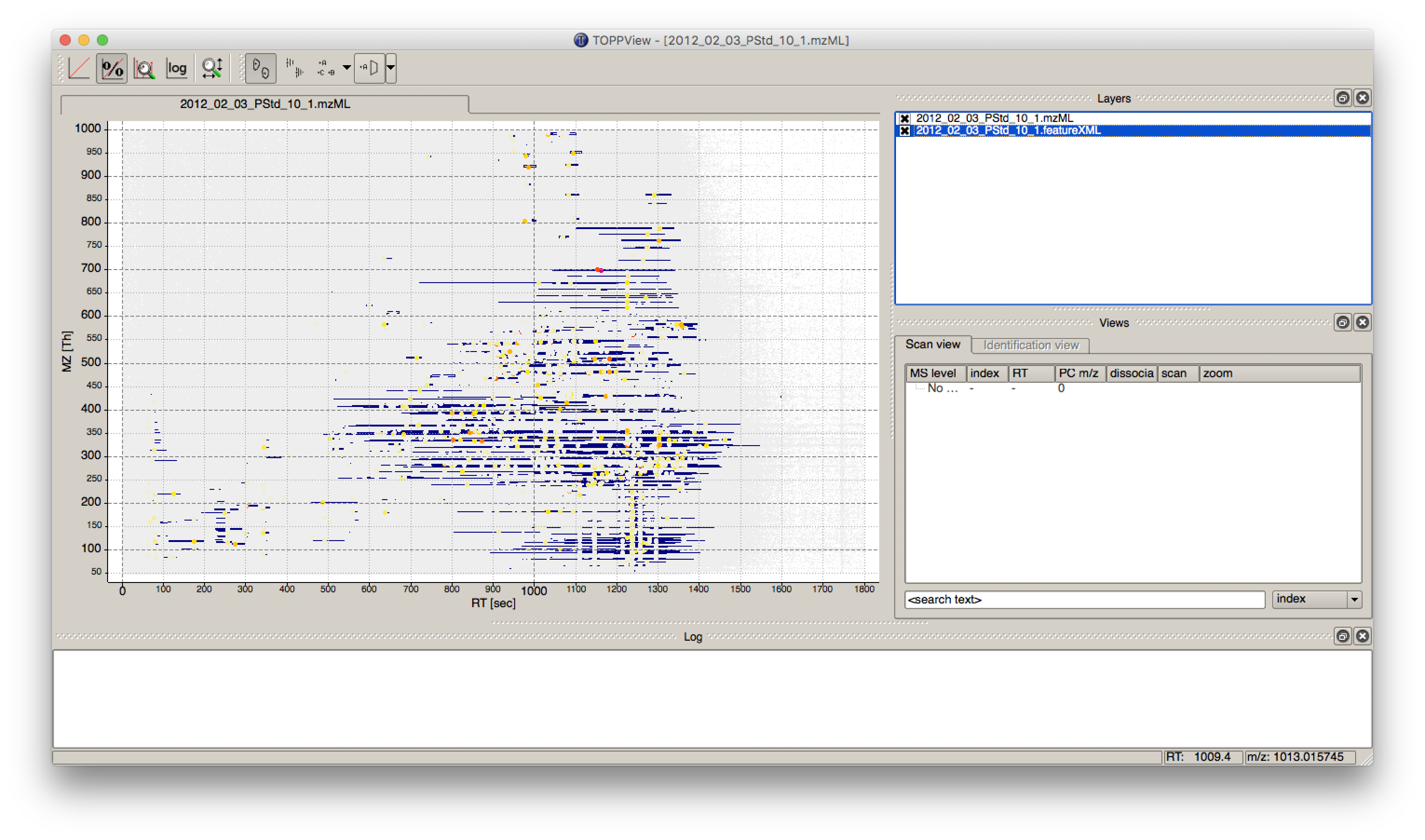
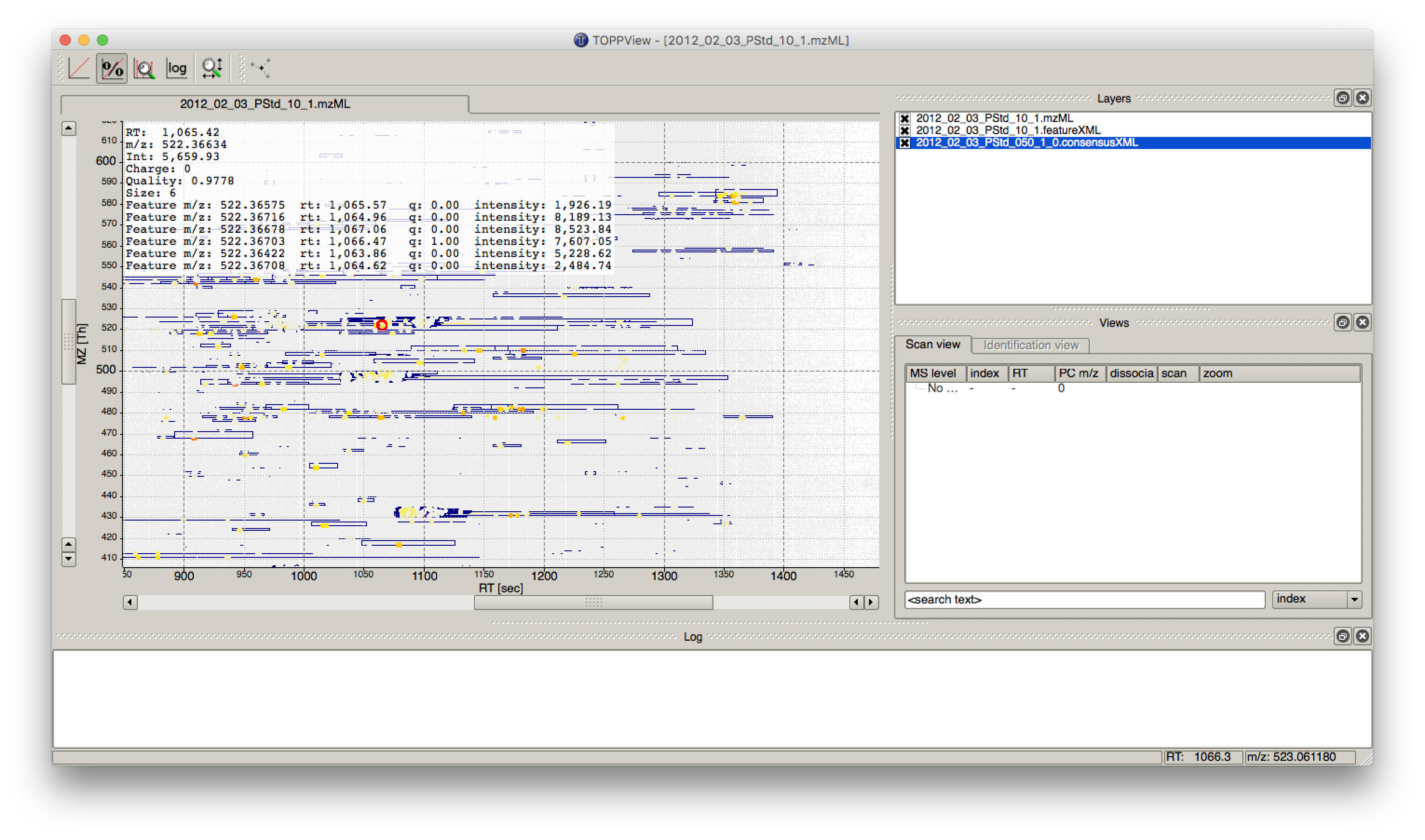
The output file .featureXML can be visualized with TOPPView on top of the used .mzML file -
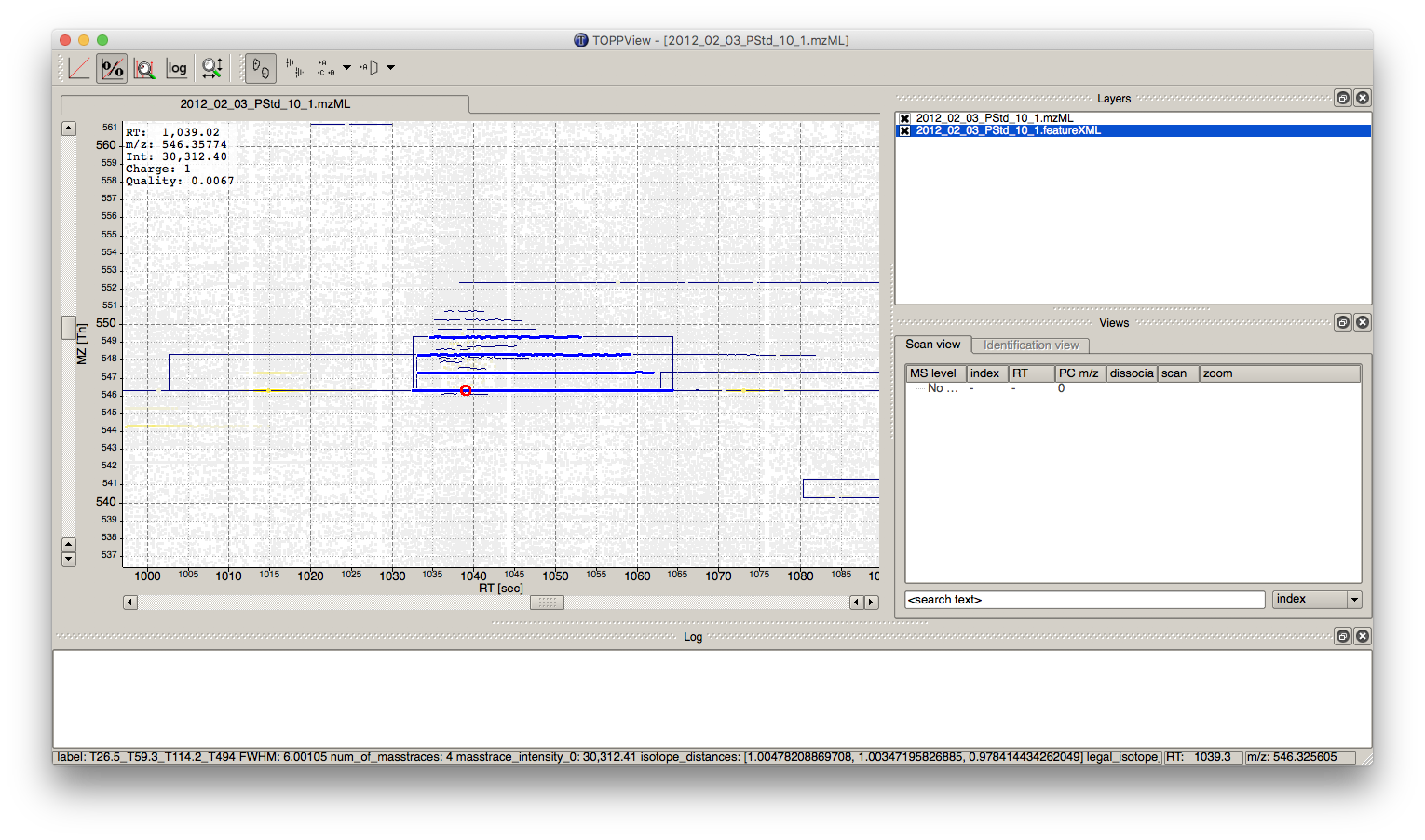
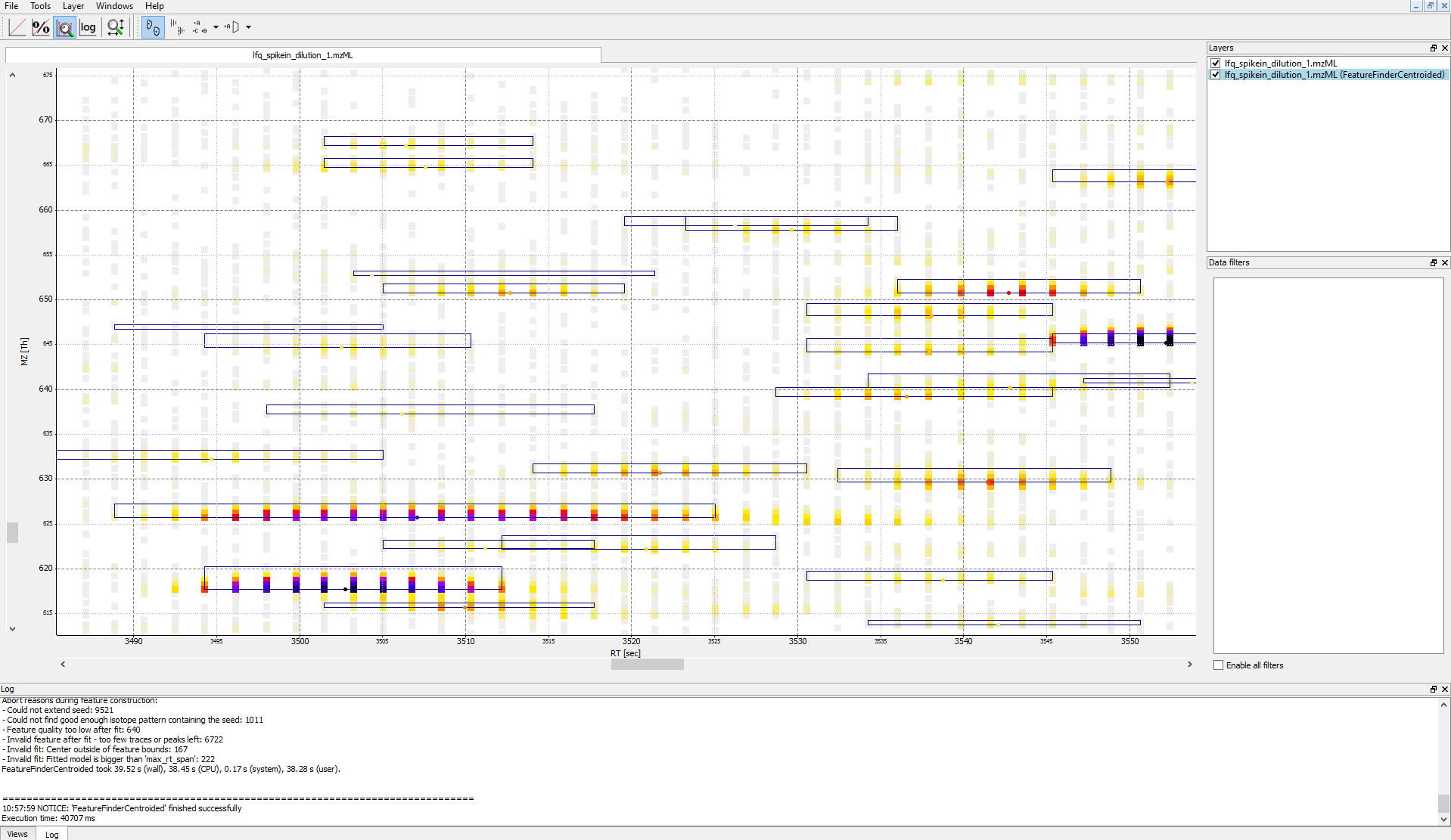
in a so called layer - to look at the identified features.
First start TOPPView and open the example .mzML file (see Fig. 28). Afterwards open the .featureXML output as new layer (see Fig. 29). The overlay is depicted in Figure 30. The zoom of the .mzML - .featureXML overlay shows the individual mass traces and the assembly of those in a feature (see Fig. 31).
The workflow can be extended for multi-file analysis, here an Input Files is to be used
instead of the Input File. In front of the FeatureFinderMetabo a ZipLoopStart and behind
ZipLoopEnd has to be used, since FeatureFinderMetabo will analyis on file to file bases.
To facilitate the collection of features corresponding to the same compound ion across
different samples, an alignment of the samples’ feature maps along retention time is
often helpful. In addition to local, small-scale elution differences, one can often see
constant retention time shifts across large sections between samples. We can use linear
transformations to correct for these large scale retention differences. This brings the majority of
corresponding compound ions close to each other. Finding the correct corresponding
ions is then faster and easier, as we don’t have to search as far around individual
features.
 ), set its Output Type to featureXML, and adjust the following settings
), set its Output Type to featureXML, and adjust the following settings
| parameter | value |
| algorithm →max_num_peaks_considered | −1 |
| algorithm →superimposer →mz_pair_max_distance | 0.005 |
| algorithm →superimposer →num_used_points | 10000 |
| algorithm →pairfinder →distance_RT →max_difference | 20.0 |
| algorithm →pairfinder →distance_MZ →max_difference | 20.0 |
| algorithm →pairfinder →distance_MZ →unit | ppm |
MapAlignerPoseClustering provides an algorithm to align the retention time scales of multiple
input files, correcting shifts and distortions between them. Retention time adjustment may be
necessary to correct for chromatography differences e.g. before data from multiple LC-MS runs
can be combined (feature linking). The alignment algorithm implemented here is the pose
clustering algorithm.
The parameters change the behavior of MapAlignerPoseClustering as follows:
The next step after retention time correction is the grouping of corresponding features in multiple samples. In contrast to the previous alignment, we assume no linear relations of features across samples. The used method is tolerant against local swaps in elution order.
 ) and adjust the following settings
) and adjust the following settings
| parameter | value |
| algorithm →distance_RT →max_difference | 40.0 |
| algorithm →distance_MZ →max_difference | 20.0 |
| algorithm →distance_MZ →unit | ppm |
The parameters change the behavior of FeatureLinkerUnlabeledQT as follows (similar to the parameters we adjusted for MapAlignerPoseClustering):
 ).
).
You should find a single, tab-separated file containing the information on where metabolites were found and with which intensities. You can also add Output Folder nodes at different stages of the workflow and inspect the intermediate results (e.g., identified metabolite features for each input map). The complete workflow can be seen in Figure 34. In the following section we will try to identify those metabolites.
The FeatureLinkerUnlabeledQT output can be visualized in ToppView on top of the input and output of the FeatureFinderMetabo (see Fig 35).
At the current state we found several metabolites in the individual maps but so far don’t know what they are. To identify metabolites OpenMS provides multiple tools, including search by mass: the AccurateMassSearch node searches observed masses against the Human Metabolome Database (HMDB)[14, 15, 16]. We start with the workflow from the previous section (see Figure 34).
 ) and connect the output of the FeatureLinkerUnlabeledQT to the incoming port.
) and connect the output of the FeatureLinkerUnlabeledQT to the incoming port.
 ) and connect the output of the FileConverter to the first port of the AccurateMassSearch.
) and connect the output of the FileConverter to the first port of the AccurateMassSearch.




The result of the AccurateMassSearch node is in the mzTab format [17] so you can easily open it in a text editor or import it into Excel or KNIME, which we will do in the next section. The complete workflow from this section is shown in Figure 36.
The result from the TextExporter node as well as the result from the AccurateMassSearch
node are files while standard KNIME nodes display and process only KNIME tables. To convert
these files into KNIME tables we need two different nodes. For the AccurateMassSearch results
we use the MzTabReader node ( )
and its Small Molecule Section port. For the result of the TextExporter we use the
ConsensusTextReader (
)
and its Small Molecule Section port. For the result of the TextExporter we use the
ConsensusTextReader ( ).
).
When executed, both nodes will import the OpenMS files and provide access to the data as
KNIME tables. The retention time values are exported as a list using the MzTabReader based
on the current PSI-Standard. This has to be parsed using the SplitCollectionColumn, which
outputs a ”Split Value 1” based on the first entry in the rention time list, which has to be
renamed to retention time using the ColumnRename. You can now combine both tables using the
Joiner node ( ) and
configure it to match the m/z and retention time values of the respective tables. The full
workflow is shown in Figure 37.
) and
configure it to match the m/z and retention time values of the respective tables. The full
workflow is shown in Figure 37.
Metabolites commonly co-elute as ions with different adducts (e.g., glutathione+H, glutathione+Na) or with charge-neutral modifications (e.g., water loss). Grouping such related ions allows to leverage information across features. For example, a low-intensity, single trace feature could still be assigned a charge and adduct due to a matching high-quality feature. Such information can then be used by several OpenMS tools, such as AccurateMassSearch, for example to narrow down candidates for identification.
For this grouping task, we provide the MetaboliteAdductDecharger node. Its method explores the combinatorial space of all adduct combinations in a charge range for optimal explanations. Using defined adduct probabilities, it assigns co-eluting features having suitable mass shifts and charges those adduct combinations which maximize overall ion probabilities.
The tool works natively with featureXML data, allowing the use of reported convex hulls. On such a single-sample level, co-elution settings can be chosen more stringently, as ionization-based adducts should not influence the elution time: Instead, elution differences of related ions should be due to slightly differently estimated times for their feature centroids.
Alternatively, consensusXML data from feature linking can be converted for use, though with less chromatographic information. Here, the elution time averaging for features linked across samples, motivates wider co-elution tolerances.
The two main tool outputs are a consensusXML file with compound groups of related input ions, and a featureXML containing the input file but annotated with inferred adduct information and charges.
Options to respect or replace ion charges or adducts allow for example:
Task
A modified metabolomics workflow with exemplary MetaboliteAdductDecharger use and
parameters is provided in . Run
the workflow, inspect tool outputs and compare AccurateMassSearch results with and without
adduct grouping.
. Run
the workflow, inspect tool outputs and compare AccurateMassSearch results with and without
adduct grouping.
Now that you have your data in KNIME you should try to get a feeling for the capabilities of KNIME.
Task
Check out the Molecule Type Cast node ( )
together with subsequent cheminformatics nodes (e.g. RDKit From Molecule (
)
together with subsequent cheminformatics nodes (e.g. RDKit From Molecule ( )) to
render the structural formula contained in the result table.
)) to
render the structural formula contained in the result table.
Task
Have a look at the Column Filter node to reduce the table to the interesting columns, e.g., only the Ids, chemical formula, and intensities.
Task
Try to compute and visualize the m/z and retention time error of the different feature elements (from the input maps) of each consensus feature. Hint: A nicely configured Math Formula (Multi Column) node should suffice.
Identifying metabolites using only the accurate mass may lead to ambiguous results. In practice, additional information (e.g. the retention time) is used to further narrow down potential candidates. Apart from MS1-based features, tandem mass spectra (MS2) of metabolites provide additional information. In this part of the tutorial, we take a look on how metabolite spectra can be identified using a library of previously identified spectra.
Because these libraries tend to be large we don’t distribute them with OpenMS.
Task
Construct the workflow as shown in Fig. 39. Use the file  as
input for your workflow. You can use the spectral library from
as
input for your workflow. You can use the spectral library from 
as second input. The first input file contains tandem spectra that are identified by the
MetaboliteSpectralMatcher. The resulting mzTab file is read back into a KNIME table The
retention time values are exported as a list based on the current PSI-Standard. This has to be
parsed using the SplitCollectionColumn, which outputs a ”Split Value 1” based on the
first entry in the rention time list, which has to be renamed to retention time using
the ColumnRename before it is stored in an Excel table. Make sure that you connect
the MzTabReader port corresponding to the Small Molecule Section to the Excel
writer (XLS). Please select the ”add column headers” option in the Excel writer
(XLS)).
Run the workflow and inspect the output.
In metabolomics, matches between tandem spectra and spectral libraries are manually validated. Several commercial and free online resources exist which help in that task. Some examples are:
Here, we will use METLIN to manually validate metabolites.
Task
Check in the .xlsx output from the Excel writer (XLS) if you can find glutathione. Use the
retention time column to find the spectrum in the mzML file. Here open the file in the
 in
TOPPView. The MSMS spectrum with the retention time of 67.6 s is used as example. The
spectrum can be selected based on the retention time in the scan view window. Therefore the
MS1 spectrum with the retention time of 66.9 s has to be double clicked and the MSMS spectra
recorded in this time frame will show up. Select the tandem spectrum of Glutathione, but do
not close TOPPView, yet.
in
TOPPView. The MSMS spectrum with the retention time of 67.6 s is used as example. The
spectrum can be selected based on the retention time in the scan view window. Therefore the
MS1 spectrum with the retention time of 66.9 s has to be double clicked and the MSMS spectra
recorded in this time frame will show up. Select the tandem spectrum of Glutathione, but do
not close TOPPView, yet.
Task
On the METLIN homepage search for  Glutathione using the
Glutathione using the  (https://metlin.scripps.edu/landing_page.php?pgcontent=advanced_search). Note that
free registration is required. Which collision energy (and polarity) gives the best (visual) match
to your experimental spectrum in TOPPView? Here you can compare the fragmentation
patterns in both spectra shown by the Intensity or relative Intensity, the m/z of a peak and the
distance between peaks. Each distance between two peaks corresponds to a fragment of
elemental composition (e.g., NH2 with the charge of one would have mass of two peaks of
16.023 Th).
(https://metlin.scripps.edu/landing_page.php?pgcontent=advanced_search). Note that
free registration is required. Which collision energy (and polarity) gives the best (visual) match
to your experimental spectrum in TOPPView? Here you can compare the fragmentation
patterns in both spectra shown by the Intensity or relative Intensity, the m/z of a peak and the
distance between peaks. Each distance between two peaks corresponds to a fragment of
elemental composition (e.g., NH2 with the charge of one would have mass of two peaks of
16.023 Th).
Another method for MS2 spectra-based metabolite identification is de novo identification. This approach can be used in addition to the other methods (accurate mass search, spectral library search) or individually if no spectral library is available. In this part of the tutorial, we discuss how metabolite spectra can be identified using de novo tools. To this end, the tools SIRIUS and CSI:FingerID ([18, 19, 20]) were integrated in the OpenMS Framework as SiriusAdapter. SIRIUS uses isotope pattern analysis to detect the molecular formula and further analyses the fragmentation pattern of a compound using fragmentation trees. CSI:FingerID is a method for searching a fingerprint of a small molecule (metabolite) in a molecular structure database.
The node is able to work in different modes depending on the provided input.
The last approach is the preferred one, as SIRIUS gains a lot of additional information by using the OpenMS tools for preprocessing.
Task
Construct the workflow as shown in Fig. 42.
Use the file  as
input for your workflow.
as
input for your workflow.
Run the workflow and inspect the output.
The output consists of two mzTab files and an internal .ms file. One mzTab for SIRIUS and the other for the CSI:FingerID. These provide information about the chemical formula, adduct and the possible compound structure. The information is referenced to the spectrum used in the analysis. Additional information can be extracted from the SiriusAdapter by setting an ”out_workspace_directory”. Here the SIRIUS workspace will be provided after the calculation has finished. This workspace contains information about annotated fragments for each successfully explained compound.
In this part of the metabolomics session we take a look at more advanced downstream analysis and the use of the statistical programming language R. As laid out in the introduction we try to detect a set of spike-in compounds against a complex blood background. As there are many ways to perform this type of analysis we provide a complete workflow.
Task
Import the workflow from  in
KNIME:
in
KNIME: 
The section below will guide you in your understanding of the different parts of the workflow. Once you understood the workflow you should play around and be creative. Maybe create a novel visualization in KNIME or R? Do some more elaborate statistical analysis? Note that some basic R knowledge is required to fully understand the processing in R Snippet nodes.
This part is analogous to what you did for the simple metabolomics pipeline.
The first part is identical to what you did for the simple metabolomics pipeline. Additionally, we convert zero intensities into NA values and remove all rows that contain at least one NA value from the analysis. We do this using a very simple R Snippet and subsequent Missing Value filter node.
Task
Inspect the R Snippet by double-clicking on it. The KNIME table that is passed to an R Snippet node is available in R as a data.frame named knime.in. The result of this node will be read from the data.frame knime.out after the script finishes. Try to understand and evaluate parts of the script (Eval Selection). In this dialog you can also print intermediary results using for example the R command head(knime.in) or cat(knime.in) to the Console pane.
After we linked features across all maps, we want to identify features that are significantly deregulated between the two conditions. We will first scale and normalize the data, then perform a t-test, and finally correct the obtained p-values for multiple testing using Benjamini-Hochberg. All of these steps will be carried out in individual R Snippet nodes.
 .
.
KNIME supports multiple nodes for interactive visualization with interrelated output. The nodes used in this part of the workflow exemplify this concept. They further demonstrate how figures with data dependent customization can be easily realized using basic KNIME nodes. Several simple operations are concatenated in order to enable an interactive volcano plot.
 ) to emphasize interesting data points. The configuration dialogs allow us to select
columns to change color, shape or size of data points dependent on the column
values.
) to emphasize interesting data points. The configuration dialogs allow us to select
columns to change color, shape or size of data points dependent on the column
values.
 ) enables interactive visualization of the logarithmized values as a volcano plot:
the log-transformed values can be chosen in the ‘Column Selection’ tab of the plot
view. Data points can be selected in the plot and highlighted via the menu option.
The highlighting transfers to all other interactive nodes connected to the same data
table. In our case, selection and the highlighting will also occur in the Interactive
Table node (
) enables interactive visualization of the logarithmized values as a volcano plot:
the log-transformed values can be chosen in the ‘Column Selection’ tab of the plot
view. Data points can be selected in the plot and highlighted via the menu option.
The highlighting transfers to all other interactive nodes connected to the same data
table. In our case, selection and the highlighting will also occur in the Interactive
Table node ( ).
).
Task
Inspect the nodes of this section. Customize your visualization and possibly try to visualize other aspects of your data.
R Dependencies: This section requires that the R packages ggplot2 and ggfortify are both installed. ggplot2 is part of the KNIME R Statistics Integration (Windows Binaries) which should already be installed via the full KNIME installer, ggfortify however is not. In case that you use an R installation where one or both of them are not yet installed, add an R Snippet node and double-click to configure. In the R Script text editor, enter the following code:
You can remove the install.packages commands once it was successfully installed.
Even though the basic capabilities for (interactive) plots in KNIME are valuable for initial data exploration, professional looking depiction of analysis results often relies on dedicated plotting libraries. The statistics language R supports the addition of a large variety of packages, including packages providing extensive plotting capabilities. This part of the workflow shows how to use R nodes in KNIME to visualize more advanced figures. Specifically, we make use of different plotting packages to realize heatmaps.
Following the identification, quantification and statistical analysis our data is merged and formatted for reporting. First we want to discard our normalized and logarithmized intensity values in favor of the original ones. To this end we first remove the intensity columns (Column Filter) and add the original intensities back (Joiner). For that we use an Inner Join 2 with the Joiner node. In the dialog of the node we add two entries for the Joining Columns and for the first column we pick ”retention_time” from the top input (i.e. the AccurateMassSearch output) and ”rt_cf” (the retention time of the consensus features) for the bottom input (the result from the quantification). For the second column you should choose ”exp_mass_to_charge” and ”mz_cf” respectively to make the joining unique. Note that the workflow needs to be executed up to the previous nodes for the possible selections of columns to appear.
Question
What happens if we use a Left Outer Join, Right Outer Join or Full Outer Join instead of the Inner Join?
Task
Inspect the output of the join operation after the Molecule Type Cast and RDKit molecular structure generation.
While all relevant information is now contained in our table the presentation could be improved. Currently, we have several rows corresponding to a single consensus feature (=linked feature) but with different, alternative identifications. It would be more convenient to have only one row for each consensus feature with all accurate mass identifications added as additional columns. To this end, we use the Column to Grid node that flattens several rows with the same consensus number into a single one. Note that we have to specify the maximum number of columns in the grid so we set this to a large value (e.g. 100). We finally export the data to an Excel file (XLS Writer).
OpenSWATH [21] allows the analysis of LC-MS/MS DIA (data independent acquisition)
data using the approach described by Gillet et al. [22]. The DIA approach described
there uses 32 cycles to iterate through precursor ion windows from 400-426 Da to
1175-1201 Da and at each step acquires a complete, multiplexed fragment ion spectrum
of all precursors present in that window. After 32 fragmentations (or 3.2 seconds),
the cycle is restarted and the first window (400-426 Da) is fragmented again, thus
delivering complete “snapshots” of all fragments of a specific window every 3.2 seconds.
The analysis approach described by Gillet et al. extracts ion traces of specific fragment ions from all MS2 spectra that have the same precursor isolation window, thus generating data that is very similar to SRM traces.
OpenSWATH has been fully integrated since OpenMS 1.10 [4, 2, 23, 24, 25]).
mProphet (http://www.mprophet.org/) [26] is available as standalone script in
 . R
(http://www.r-project.org/) and the package MASS (http://cran.r-project.org/web/packages/MASS/)
are further required to execute mProphet. Please obtain a version for either Windows, Mac or
Linux directly from CRAN.
. R
(http://www.r-project.org/) and the package MASS (http://cran.r-project.org/web/packages/MASS/)
are further required to execute mProphet. Please obtain a version for either Windows, Mac or
Linux directly from CRAN.
PyProphet, a much faster reimplementation of the mProphet algorithm is available from PyPI
(https://pypi.python.org/pypi/pyprophet/). The usage of pyprophet instead of mProphet
is suggested for large-scale applications.
mProphet will be used in this tutorial.
OpenSWATH requires an assay library to be supplied in the TraML format [27]. To
enable manual editing of transition lists, the TOPP tool TargetedFileConverter is
available, which uses tab separated files as input. Example datasets are provided in
 .
Please note that the transition lists need to be named .tsv.
.
Please note that the transition lists need to be named .tsv.
The header of the transition list contains the following variables (with example values in brackets):
For further instructions about generic transition list and assay library generation please see
http://openswath.org/en/latest/docs/generic.html.
To convert transitions lists to TraML, use the TargetedFileConverter: Please use the absolute
path to your OpenMS installation.
In addition to the target assays, OpenSWATH requires decoy assays in the library which are later used for classification and error rate estimation. For the decoy generation it is crucial that the decoys represent the targets in a realistic but unnatural manner without interfering with the targets. The methods for decoy generation implemented in OpenSWATH include ’shuffle’, ’pseudo-reverse’, ’reverse’ and ’shift’. To append decoys to a TraML, the TOPP tool OpenSwathDecoyGenerator can be used: Please use the absolute path to your OpenMS installation.
An example KNIME workflow for OpenSWATH is supplied in  (Fig. 44). The example dataset can be used for this workflow (filenames in brackets):
(Fig. 44). The example dataset can be used for this workflow (filenames in brackets):
 in KNIME:
in KNIME:  .
.
 .
. )
)
 .
. )
)
 .
. )
)
 .
.The resulting output can be found at your selected path, which will be used as input for
mProphet. Execute the script on the Terminal (Linux or Mac) or cmd.exe (Windows) in
 .
Please use the absolute path to your R installation and the result file:
.
Please use the absolute path to your R installation and the result file:
or for windows
The main output will be called
with statistical information available in .
.
Please note that due to the semi-supervised machine learning approach of mProphet the results
differ slightly when mProphet is executed several times.
Additionally the chromatrogam output (.mzML) can be visualized for inspection with
TOPPView.
For additional instructions on how to use pyProphet instead of mProphet please have a look at the
PyProphet Legacy Workflow http://openswath.org/en/latest/docs/pyprophet_legacy.html.
If you want to use the SQLite-based workflow in your lab in the future, please have a look here:
http://openswath.org/en/latest/docs/pyprophet.html. The SQLite-based workflow will
not be part of the tutorial.
The sample dataset used in this tutorial is part of the larger SWATH MS Gold Standard (SGS) dataset which is described in the publication of Roest et al. [21]. It contains one of 90 SWATH-MS runs with significant data reduction (peak picking of the raw, profile data) to make file transfer and working with it easier. Usually SWATH-MS datasets are huge with several gigabyte per run. Especially when complex samples in combination with large assay libraries are analyzed, the TOPP tool based workflow requires a lot of computational resources. Additional information and instruction can be found at http://openswath.org/en/latest/.
We would like to present an automated DIA/SWATH analysis workflow for metabolomics, which takes advantage of experiment specific target-decoy assay library generation. This allows for targeted extraction, scoring and statistical validation of metabolomics DIA data [29], [30].
The workflow follows multiple steps (see Fig. 45).
Candidate identification Feature detection, adduct grouping and accurate mass search are applied on DDA data.
Library construction The knowledge determined from the DDA data, about compound identification, its potential adduct and the corresponding fragment spectra are used to perform fragment annotation via compositional fragmentation trees [31]. Afterwards transitions, which are the reference of a precursor to its fragment ions are stored in a so-called assay library. Assay libraries usually contain additional metadata (i.e. retention time, peak intensities). FDR estimation is based on the target-decoy approach [32]. For the generation of the MS2 decoys, the fragmentation tree-based rerooting method ensures the consistency of decoy spectra [33]. Targeted extraction - The target-decoy assay library is then used to analyse SWATH data.
Targeted extraction Chromatogram extraction and peak-group scoring. This step is performed using an algorithm based on OpenSWATH [29] for metabolomics data.
Statistical validation FDR estimation uses the PyProphet algorithm [30]. To prevent overfitting we chose the simpler linear model (LDA) for target-decoy discrimination in PyProphet, using MS1 and MS2 scoring with low correlated scores.
Apart from the usual KNIME nodes, the workflow uses python scripting nodes. One basic
requirement for the installation of python packages, in particular pyOpenMS, is a package
manager for python. Using conda as an environment manger allows to specify a specific
environment in the KNIME settings ( )
)
We suggest do use a virtual environment for the Python 3 installation on windows. Here you
can install miniconda and follow the further instructions.
We suggest do use a virtual environment for the Python 3 installation on Mac. Here you can
install miniconda and follow the further instructions.
Use your package manager apt-get or yum, where possible.
For the assay library construction pesticide mixes (Agilent Technologies, Waldbronn, Germany) were measured individually in solvent (DDA). Benchmark DIA samples were prepared by spiking different commercially available pesticide mixes into human plasma metabolite extracts in a 1:4 dilution series, which covers 5 orders of magnitude.
The example data can be found here: https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Tutorials/Data/DIAMetAlyzer/
Example workflow for the usage of the DIAMetAlyzer Pipeline in KNIME (see Fig. 46). Inputs are the SWATH-MS data in profile mode (.mzML), a path for saving the new target-decoy assay library, the SIRIUS 4.0.1 executable, the DDA data (.mzML), custom libraries and adducts for AccurateMassSearch, the min/max fragment mass-to-charge to be able to restrict the mass of the transitions and the path to the PyProphet executable. The DDA is used for feature detection, adduct grouping, accurate mass search and forwarded to the AssayGeneratorMetabo. The constructed target library is used by the decoy generation node, which calls various python scripts to parse, reformat the input, call Passatutto and export the target-decoy assay library. The target-decoy assay library is processed with the SWATH-MS data in OpenSWATH. The results are used by PyProphet for scoring and output a list of metabolites with their respective q-value and quantitative information.
These steps need to be followed to run the workflow successfully:
Please have a look at the most important parameters, which should be tweaked to fit your data.
In general, OpenMS has a lot of room for parameter optimization to best fit your
chromatography and instrumental settings.
FeatureFinderMetabo:
| parameter | explanation |
| noise_threshold_int | Intensity threshold below which peaks are regarded as noise. |
| chrom_fwhm | Expected chromatographic peak width (in seconds). |
| mass_error_ppm | Allowed mass deviation (in ppm) |
MetaboliteAdductDecharger:
| parameter | explanation |
| mass_max_diff | Maximum allowed mass tolerance per feature.. |
| potential_adducts | Adducts used to explain mass differences - These should fit to the adduct list specified for AccurateMassSearch. |
AccurateMassSearch:
| parameter | explanation |
| mass_error_value | Tolerance allowed for accurate mass search. |
| ionization_mode | Positive or negative ionization mode. |
AssayGeneratorMetabo:
| parameter | explanation |
| min_transitions | Minimal number of transitions (3). |
| max_transitions | Maximal number of transitions (3). |
| transitions_threshold | Further transitions need at least x% of the maximum intensity. |
| out_workspace_directory | Output directory for SIRIUS workspace (Fragmentation Trees). |
| filter_by_num_masstraces | Features have to have at least x MassTraces. To use this parameter feature_only is neccessary. |
| precursor_mass_tolerance | Tolerance window for precursor selection (Feature selection in regard to the precursor). |
| precursor_rt_tolerance | Tolerance allowed for matching MS2 spectra depeding on the feature size (should be around the FWHM of the chromatograms). |
| profile | Specify the used analysis profile (e.g. qtof). |
| elements | Allowed elements for assessing the putative sumformula (e.g. CHNOP[5]S[8]Cl[1]). Elements found in the isotopic pattern are added automatically, but can be specified nonetheless. |
OpenSWATH:
| parameter | explanation |
| rt_extraction_window | Extract x seconds around this value. |
| rt_normalization_factor | Please use the range of your gradient e.g. 950 seconds. |
If you are analysing a lot of big DIA mzML files ≈ 3-20GB per File, it makes sense to change how OpenSWATH processes the spectra.
| parameter | explanation |
| readOptions | Set cacheWorkingInMemory - will cache the files to disk and read SWATH-by-SWATH into memory |
| tempDirectory | Set a directory, where cached mzMLs are stored (be aware that his directory can be quite huge depending on the data). |
In the workflow pyprophet is called after OpenSWATH, it merges the result files, which allows to get enough data for the model training.
Afterwards, the results are scored using the MS1 and MS2 levels and filter for metabolomics scores, which have a low correlation.
Export the non filtered results:
Please see the workflow for actual parameter values used for the benchmarking dataset.
The workflow can be used without any identification (remove AccurateMassSearch). Here, all features (known_unknowns) are processed. The assay library is constructed based on the chemical composition elucidated via the fragment annotation. Quantification is possible, unfortunately the decoy generation and FDR estimation for this case is not yet available.
pyOpenMS provides Python bindings for a large part of the OpenMS library for mass
spectrometry based proteomics and metabolomics. It thus provides access to a feature-rich,
open-source algorithm library for mass-spectrometry based LC-MS analysis. These
Python bindings allow raw access to the data-structures and algorithms implemented in
OpenMS, specifically those for file access (mzXML, mzML, TraML, mzIdentML among
others), basic signal processing (smoothing, filtering, de-isotoping and peak-picking) and
complex data analysis (including label-free, SILAC, iTRAQ and SWATH analysis
tools).
pyOpenMS is integrated into OpenMS starting from version 1.11. This tutorial is addressed to people already familiar with Python. If you are new to Python, we suggest to start with a Python tutorial (https://en.wikibooks.org/wiki/Non-Programmer%27s_Tutorial_for_Python_3).
One basic requirement for the installation of python packages, in particular pyOpenMS, is a package manager for python. We provide a package for pip (https://pypi.python.org/pypi/pip).
We suggest do use a virtual environment for the Python 3 installation on Mac. Here you can
install miniconda and follow the further instructions.
Use your package manager apt-get or yum, where possible.
If you do not have python installed or do not want to modify your native installation, another
possibility is to use an IDE (integrated development environment) with Anaconda
integration. Here, we recommend spyder (https://www.spyder-ide.org/). It comes with
Anaconda, which is a package and environment manager. Thus the IDE should be
able to run a specific environment independent of your systems python installation.
Please execute the installer for your respective platform located in the respective directory for
your platform and follow the installation instructions.
After installation the ANACONDA Navigator (Anaconda 3) should be available. Please start
the application. To install pyopenms please choose the button ”Environments” and click the
play symbol of the base environment and ”Open Terminal”.
Update pip and install pyopenms (MacOS, Linux):
Update pip and install pyopenms (Windows):
Install a local available package:
The local available packages can be found in the directory corresponding to your operating system. Please use the absolute path to the packages for the installation.
Now launch ”Spyder” (python IDE) in the home menu.
Instructions on how to build pyOpenMS can be found online (https://pyopenms.readthedocs.io/en/release_2.4.0/build_from_source.html).
A big advantage of pyOpenMS are its scripting capabilities (beyond its application in
tool development). Most of the OpenMS datastructure can be accessed using python
(https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/index.html).
Here we would like to give some examples on how pyOpenMS can be used for simple scripting
task, such as peptide mass calculation and peptide/protein digestion as well as isotope
distribution calculation.
Calculation of the monoisotopic and average mass of a peptide sequence
from pyopenms import * seq = AASequence.fromString("DFPIANGER") mono_mass = seq.getMonoWeight(Residue.ResidueType.Full, 0) average_mass = seq.getAverageWeight(Residue.ResidueType.Full, 0) print("The masses of the peptide sequence " + seq.toString().decode('utf-8') + " are:") print("mono: " + str(mono_mass)) print("average: "+ str(average_mass))
Enzymatic digest of a peptide/protein sequence
enzyme = "Trypsin" to_digest = AASequence.fromString("MKWVTFISLLLLFSSAYSRGVFRRDTHKSEIAHRFKDLGE") after_digest = [] EnzymaticDigest = EnzymaticDigestionLogModel() EnzymaticDigest.setEnzyme(enzyme) EnzymaticDigest.digest(to_digest, after_digest) print("The peptide " + to_digest.toString().decode('utf-8') + " was digested using " + str(EnzymaticDigest.getEnzymeName().decode('utf-8')) + " to:") for element in after_digest: print(element.toString().decode('utf-8'))
Use empirical formula to calculate the isotope distribution
from pyopenms import * methanol = EmpiricalFormula("CH3OH") water = EmpiricalFormula("H2O") wm = EmpiricalFormula(water.toString().decode('utf-8') + methanol.toString().decode('utf-8')) print(wm.toString().decode('utf-8')) print(wm.getElementalComposition()) isotopes = wm.getIsotopeDistribution( CoarseIsotopePatternGenerator(3) ) for iso in isotopes.getContainer(): print (iso.getMZ(), ":", iso.getIntensity())
For further examples and the pyOpenMS datastructure please see https://pyopenms.readthedocs.io/en/release_2.4.0/datastructures.html.
Scripting is one side of pyOpenMS, the other is the ability to create Tools using the
C++ OpenMS library in the background. In the following section we will create a
”ProteinDigestor” pyOpenMS Tool. It should be able to read in a fasta file. Digest the proteins
with a specific enzyme (e.g. Trypsin) and export an idXML output file. Please see
 for
code snippets.
for
code snippets.
usage: ProteinDigestor.py [-h] [-in INFILE] [-out OUTFILE] [-enzyme ENZYME] [-min_length MIN_LENGTH] [-max_length MAX_LENGTH] [-missed_cleavages MISSED_CLEAVAGES] ProteinDigestor −− In silico digestion of proteins. optional arguments: -h, --help show this help message and exit -in INFILE An input file containing amino acid sequences [fasta] -out OUTFILE Output digested sequences in idXML format [idXML] -enzyme ENZYME Enzyme used for digestion -min_length MIN_LENGTH Minimum length of peptide -max_length MAX_LENGTH Maximum length of peptide -missed_cleavages MISSED_CLEAVAGES The number of allowed missed cleavages
First, your tool needs to be able to read parameters from the command line and provide a main routine. Here standard Python can be used (no pyOpenMS is required so far).
#!/usr/bin/env python import sys def main(options): # test parameter handling print(options.infile, options.outfile, options.enzyme, options.min_length, options.max_length, options.missed_cleavages) def handle_args(): import argparse usage = "" usage += "\nProteinDigestor −− In silico digestion of proteins." parser = argparse.ArgumentParser(description = usage) parser.add_argument('-in', dest='infile', help='An input file containing amino acid sequences [fasta]') parser.add_argument('-out', dest='outfile', help='Output digested sequences in idXML format [idXML]') parser.add_argument('-enzyme', dest='enzyme', help='Enzyme used for digestion') parser.add_argument('-min_length', type=int, dest='min_length', help ='Minimum length of peptide') parser.add_argument('-max_length', type=int, dest='max_length', help='Maximum length of peptide') parser.add_argument('-missed_cleavages', type=int, dest='missed_cleavages', help='The number of allowed missed cleavages') args = parser.parse_args(sys.argv[1:]) return args if __name__ == '__main__': options = handle_args() main(options)
Open the Anaconda Terminal and change into the  directory. Execute the example script.
directory. Execute the example script.
The parameters are being read from the command line by the function handle_args() and given to the main() function of the script, which prints the different variables.
OpenMS has a ProteaseDB class containing a list of enzymes which can be used for digestion of proteins. You can add this to the argparse code to be able to see the usable enzymes. From this point onward pyOpenMS is required.
# from here pyopenms is needed # get available enzymes from ProteaseDB all_enzymes = [] p_db=ProteaseDB().getAllNames(all_enzymes) # concatenate them to the enzyme argument. parser.add_argument('-enzyme', dest='enzyme', help='Enzymes which can be used for digestion: '+ ', '.join(map(bytes.decode, all_enzymes)))
We already scripted enzymatic digestion with the AASequence and EnzymaticDigest (see above). To make this even easier, we can use an existing class in OpenMS, called ProteaseDigestion.
# Use the ProteaseDigestion class # set the enzyme used for digestion and the number of missed cleavages digestor = ProteaseDigestion() digestor.setEnzyme(options.enzyme) digestor.setMissedCleavages(options.missed_cleavages) # call the ProteaseDigestion::digest function # which will return the number of discarded digestions products # and fill the current_digest list with digestes peptide sequences digestor.digest(aaseq.fromString(fe.sequence), current_digest, options.min_length, options.max_length)
The next step is to use FASTAFile class to read the fasta input:
The output idXML needs the information about protein and peptide level, which can be saved in the ProteinIdentification and PeptideIdentification classes.
This is the part of the program which unifies the snippets provided above. Please have a closer look how the protein and peptide datastructure is incorporated in the program.
def main(options): # read fasta file ff = FASTAFile() ff.readStart(options.infile) fe = FASTAEntry() # use ProteaseDigestion class digestor = ProteaseDigestion() digestor.setEnzyme(options.enzyme) digestor.setMissedCleavages(options.missed_cleavages) # protein and peptide datastructure protein_identifications = [] peptide_identifications = [] protein_identification = ProteinIdentification() protein_identifications.append(protein_identification) temp_pe = PeptideEvidence() # number of dropped peptides due to length restriction dropped_by_length = 0 while(ff.readNext(fe)): # construct ProteinHit and fill it with sequence information temp_protein_hit = ProteinHit() temp_protein_hit.setSequence(fe.sequence) temp_protein_hit.setAccession(fe.identifier) # save the ProteinHit in a ProteinIdentification Object protein_identification.insertHit(temp_protein_hit) # construct a PeptideHit and save the ProteinEvidence (Mapping) for the specific # current protein temp_peptide_hit = PeptideHit() temp_pe.setProteinAccession(fe.identifier); temp_peptide_hit.setPeptideEvidences([temp_pe]) # digestion current_digest = [] aaseq = AASequence() if (options.enzyme == "none"): current_digest.append(aaseq.fromString(fe.sequence)) else: dropped_by_length += digestor.digest(aaseq.fromString(fe.sequence), current_digest, options.min_length, options.max_length) for seq in current_digest: # fill the PeptideHit and PeptideIdentification datastructure peptide_identification = PeptideIdentification() temp_peptide_hit.setSequence(seq) peptide_identification.insertHit(temp_peptide_hit) peptide_identifications.append(peptide_identification) print(str(dropped_by_length) + " peptides have been dropped due to the length restriction.") idxml = IdXMLFile() idxml.store(options.outfile, protein_identifications, peptide_identifications)
The paramter input and the functions can be used to construct the program we are looking for. If you are struggling please have a look in the example data section ProteinDigestor.py
Now you can run your tool in the Anaconda Terminal (  ):
):
Task
Implement all other 184 TOPP tools using pyOpenMS.
In this chapter, we will build on an existing workflow with OpenMS / KNIME to add some
quality control (QC). We will utilize the qcML tools in OpenMS to create a file with which we
can collect different measures of quality to the mass spectrometry runs themselves and the
applied analysis. The file also serves the means of visually reporting on the collected quality
measures and later storage along the other analysis result files. We will, step-by-step, extend the
label-free quantitation workflow from section 3 with QC functions and thereby enrich each
time the report given by the qcML file. But first, to make sure you get the most of
this tutorial section, a little primer on how we handle QC on the technical level.
To assert the quality of a measurement or analysis we use quality metrics. Metrics are
describing a certain aspect of the measurement or analysis and can be anything from a single
value, over a range of values to an image plot or other summary. Thus, qcML metric
representation is divided into QC parameters (QP) and QC attachments (QA) to be able to
represent all sorts of metrics on a technical level.
A QP may (or may not) have a value which would equal a metric describable with a
single value. If the metric is more complex and needs more than just a single value,
the QP does not require the single value but rather depends on an attachment of
values (QA) for full meaning. Such a QA holds the plot or the range of values in
a table-like form. Like this, we can describe any metric by a QP and an optional
QA.
To assure a consensual meaning of the quality parameters and attachments, we created a
controlled vocabulary (CV). Each entry in the CV describes a metric or part/extension thereof.
We embed each parameter or attachment with one of these and by doing so, connect a
meaning to the QP/QA. Like this, we later know exactly what we collected and the
programs can find and connect the right dots for rendering the report or calculating new
metrics automatically. You can find the constantly growing controlled vocabulary
here: .
.
Finally, in a qcml file, we split the metrics on a per mass-spectrometry-run base or a set of
mass-spectrometry-runs respectively. Each run or set will contain its QP/QA we calculate for it,
describing their quality.
As a start, we will build a basic qcML file for each mzML file in the label-free analysis. We are
already creating the two necessary analysis files to build a basic qcML file upon each mzML
file, a feature file and an identification file. We use the QCCalculator node from
 where also all other QC* nodes will be found. The QCCalculator will create a very basic qcML
file in which it will store collected and calculated quality data.
where also all other QC* nodes will be found. The QCCalculator will create a very basic qcML
file in which it will store collected and calculated quality data.
The created qcML files will not have much to show for, basic as they are. So we will extend them with some basic plots.
 . Change the width and heigth parameters to 640x640 as we don’t want it to be too
big. Connect it to the first ZipLoopStart outlet port, so it will create an image file
of the mzML’s contained run.
. Change the width and heigth parameters to 640x640 as we don’t want it to be too
big. Connect it to the first ZipLoopStart outlet port, so it will create an image file
of the mzML’s contained run.
 .
.There are two other basic plots which we almost always might want to look at before judging the quality of a mass spectrometry run and its identifications: the total ion current (TIC) and the PSM mass error (Mass accuracy), which we have available as pre-packaged QC metanodes.
Task
Import the workflow from  in
KNIME:
in
KNIME: 
R Dependencies: This section requires that the R packages ggplot2 and scales are both
installed. This is the same procedure as in section 6.4.5. In case that you use an R installation
where one or both of them are not yet installed, open the R Snippet nodes inside the
metanodes you just used (double-click). Edit the script in the R Script text editor
from:
to
Press  to
execute the script.
to
execute the script.
Note: To have a peek into what our qcML now looks like for one of the ZipLoop iterations, we
can add an Output Folder node from  and
set its destination parameter to somewhere we want to find our intermediate qcML files in, for
example
and
set its destination parameter to somewhere we want to find our intermediate qcML files in, for
example  . If
we now connect the last metanode with the Output Folder and restart the workflow, we can
start inspecting the qcML files.
. If
we now connect the last metanode with the Output Folder and restart the workflow, we can
start inspecting the qcML files.
Task
Find your first created qcML file and open it with the browser (not IE), and the contained QC parameters will be rendered for you.
We can also add brand new QC metrics to our qcML files. Remember the Histogram you added inside the ZipLoop during the label-free quantitation section? Let’s imagine for a moment this was a brand new and utterly important metric and plot for the assessment of your analyses quality. There is an easy way to integrate such new metrics into your qcMLs. Though the Histogram node cannot pass its plot to an image, we can do so with a R View (table).
 to the R View (table).
to the R View (table).
 , we found that we can attach our plot to the MS identification result details by
setting the parameter qp_att_acc to QC:0000025, as we are plotting the charge
histogram of our identified peptides.
, we found that we can attach our plot to the MS identification result details by
setting the parameter qp_att_acc to QC:0000025, as we are plotting the charge
histogram of our identified peptides.
Besides monitoring the quality of each individual mass spectrometry run analysis, another
capability of QC with OpenMS and qcML is to monitor the complete set. The easiest control is
to compare mass spectrometry runs which should be similar, e.g. technical replicates, to spot
any aberrations in the set.
For this, we will first collect all created qcML files, merge them together and use the qcML
onboard set QC properties to detect any outliers.
When inspecting the set-qcML file in a browser, we will be presented another overview. After the set content listing, the basic QC parameters (like number of identifications) are each displayed in a graph. Each set member (or run) has its own section on the x-axis and each run is connected with that graph via a link in the mouseover on one of the QC parameter values.
Task
For ideas on new QC metrics and parameters -as you add them in your qcML files as generic parameters, feel free to contact us, so we can include them in the CV.
This section will show you where you can turn to when you encounter any problems with this tutorial or with our nodes in general. Please see the FAQ first. If your problem is not listed or the proposed solution does not work, feel free to leave us a message at the means of support that you see most fit. If that is the case, please provide us with as much information as you can. In an ideal case, that would be:

 . This shows you the output of the OpenMS executable that was called by that
node. For advanced users, you can try to execute the underlying executable in your
. This shows you the output of the OpenMS executable that was called by that
node. For advanced users, you can try to execute the underlying executable in your folder, to see if the error is reproducible outside of KNIME.
folder, to see if the error is reproducible outside of KNIME.
Q: Can I add my own modifications to the Unimod.xml?
A: Unfortunately not very easy. This is an open issue since the selections are hard-coded during
creation of the tools. We included 10 places for dummy modifications that can be entered in our
Unimod.xml and selected in KNIME.
Q: I have problem XYZ but it also occurs with other nodes or generally in the KNIME
environment/GUI, what should I do?
A: This sounds like a general KNIME bug and we advise to search help directly at the KNIME
developers. They also provide a FAQ and a forum.
Q: After exporting and reading in results into a KNIME table (e.g. with a MzTabExporter and
MzTabReader combination) numeric values get rounded (e.g. from scientific notation
4.5e-10 to zero) or are in a different representation than in the underlying exported
file!
A: Please try a different table column renderer in KNIME. Open the table in question,
right-click on the header of an affected column and select another Available Renderer by
hovering and finally left-clicking.
Q: I have checked all the configurations but KNIME complains that it can not find certain
output Files (FileStoreObjects).
A: Sometimes KNIME/GKN has hiccups with multiple nodes with a same name, executed at
the same time in the same loop. We have seen that a simple save and restart of KNIME usually
solves the problem.
Linux
Q: Whenever I try to execute an OpenMS node I get an error similar to these:
/usr/lib/x86_64-linux-gnu/libgomp.so.1: version `GOMP_4.0' not found /usr/lib/x86_64-linux-gnu/libstdc++.so.6: version `GLIBCXX_3.4.20' not found
A: We currently build the binaries shipped in the OpenMS KNIME plugin with gcc 4.8. We
will try to extend our support for older compilers. Until then you either need to upgrade your
gcc compiler or at least the library that the tool complained about or you need to build the
binaries yourself (see OpenMS documentation) and replace them in your KNIME binary
folder
( ).
).
Q: Why is my configuration dialog closing right away when I double-click or try to configure it?
Or why is my GUI responding so slow?
A: If you have any problems with the KNIME GUI or the opening of dialogues under Linux you
might be affected by a GTK bug. See the KNIME forum (e.g. here or here) for a
discussion and a possible solution. In short: set environment variable by calling export
SWT_GTK3=0 or edit knime.ini to make Eclipse use GTK2 by adding the following two
lines:
–launcher.GTK_version
2
macOS
Q: I have problems installing RServe in my local R installation for the R KNIME
Extension:
A: If you encounter linker errors while running install.packages(”Rserve”) when using an R
installation from homebrew, make sure gettext is installed via homebrew and you pass flags to
its lib directory. See StackOverflow question 21370363.
Q: Although I  +
+ TOPPAS.app or TOPPView.app and accept the risk of a downloaded application, the icon only
shortly blinks and nothing happens:
TOPPAS.app or TOPPView.app and accept the risk of a downloaded application, the icon only
shortly blinks and nothing happens:
A: It seems like your OS is not able to remove the quarantine flag. If you trust us, please
remove it yourself by typing the following command in your Terminal.app:
Windows
Q: KNIME has problems getting the requirements for some of the OpenMS nodes on Windows,
what can I do?
A: Get the prerequisites installer here or install .NET3.5, .NET4 and VCRedist10.0 and 12.0
yourself.
Q: Why is my XTandemAdapter printing empty or VERY few results, although I did not use
an e-value cutoff?
A: Due to a bug in OpenMS 2.0.1 the XTandemAdapter requires a default parameter file. Give
it the default configuration in
 as a
third input file. This should be resolved in newer versions though, such that it automatically
uses this file if the optional inputs is empty. This should be solved in newer versions.
as a
third input file. This should be resolved in newer versions though, such that it automatically
uses this file if the optional inputs is empty. This should be solved in newer versions.
Q: Do MSGFPlusAdapter, LuciphorAdapter or SiriusAdapter generally behave
different/unexpected?
A: These are Java processes that are started underneath. For example they can not be killed
during cancellation of the node. This should not affect its performance, however. Make
sure you set the Java memory parameter in these nodes to a reasonable value. Also
MSGFPlus is creating several auxiliary files and accesses them during execution. Some
users therefore experienced problems when executing several instances at the same
time.
If your questions could not be answered by the FAQ, please feel free to turn to our developers via one of the following means:
[1] OpenMS, OpenMS home page [online].
[2] M. Sturm, A. Bertsch, C. Gröpl, A. Hildebrandt, R. Hussong, E. Lange, N. Pfeifer, O. Schulz-Trieglaff, A. Zerck, K. Reinert, and O. Kohlbacher, OpenMS - an open-source software framework for mass spectrometry., BMC bioinformatics 9(1) (2008), doi:10.1186/1471-2105-9-163.
[3] H. L. Röst, T. Sachsenberg, S. Aiche, C. Bielow, H. Weisser, F. Aicheler, S. Andreotti, H.-C. Ehrlich, P. Gutenbrunner, E. Kenar, et al., OpenMS: a flexible open-source software platform for mass spectrometry data analysis, Nature Methods 13(9), 741–748 (2016).
[4] O. Kohlbacher, K. Reinert, C. Gröpl, E. Lange, N. Pfeifer, O. Schulz-Trieglaff, and M. Sturm, TOPP–the OpenMS proteomics pipeline., Bioinformatics 23(2) (Jan. 2007).
[5] M. R. Berthold, N. Cebron, F. Dill, T. R. Gabriel, T. Kötter, T. Meinl, P. Ohl, C. Sieb, K. Thiel, and B. Wiswedel, KNIME: The Konstanz Information Miner, in Studies in Classification, Data Analysis, and Knowledge Organization (GfKL 2007), Springer, 2007.
[6] M. Sturm and O. Kohlbacher, TOPPView: An Open-Source Viewer for Mass Spectrometry Data, Journal of proteome research 8(7), 3760–3763 (July 2009), doi:10.1021/pr900171m.
[7] RDKit: Open-source cheminformatics, http://www.rdkit.org, [Online; accessed 31-August-2018].
[8] C. Steinbeck, Y. Han, S. Kuhn, O. Horlacher, E. Luttmann, and E. Willighagen, The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics, Journal of Chemical Information and Computer Sciences 43(2), 493–500 (2003), PMID: 12653513, doi:10.1021/ci025584y.
[9] L. Y. Geer, S. P. Markey, J. A. Kowalak, L. Wagner, M. Xu, D. M. Maynard, X. Yang, W. Shi, and S. H. Bryant, Open mass spectrometry search algorithm, Journal of Proteome Research 3(5), 958–964 (2004).
[10] A. Chawade, M. Sandin, J. Teleman, J. Malmström, and F. Levander, Data Processing Has Major Impact on the Outcome of Quantitative Label-Free LC-MS Analysis, Journal of Proteome Research 14(2), 676–687 (2015), PMID: 25407311, arXiv:http://dx.doi.org/10.1021/pr500665j, doi:10.1021/pr500665j.
[11] M. Choi, C.-Y. Chang, T. Clough, D. Broudy, T. Killeen, B. MacLean, and O. Vitek, MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments, Bioinformatics 30(17), 2524–2526 (2014), doi:10.1093/bioinformatics/btu305.
[12] M. Choi, Z. F. Eren-Dogu, C. Colangelo, J. Cottrell, M. R. Hoopmann, E. A. Kapp, S. Kim, H. Lam, T. A. Neubert, M. Palmblad, B. S. Phinney, S. T. Weintraub, B. MacLean, and O. Vitek, ABRF Proteome Informatics Research Group (iPRG) 2015 Study: Detection of Differentially Abundant Proteins in Label-Free Quantitative LC-MS/MS Experiments, J. Proteome Res. 16(2), 945–957 (2017), doi:10.1021/acs.jproteome.6b00881.
[13] T. Huang, M. Choi, S. Hao, and O. Vitek, MSstatsTMT: Protein Significance Analysis in shotgun mass spectrometry-based proteomic experiments with tandem mass tag (TMT) labeling., (2020), doi:10.18129/B9.bioc.MSstatsTMT.
[14] D. S. Wishart, D. Tzur, C. Knox, et al., HMDB: the Human Metabolome Database, Nucleic Acids Res 35(Database issue), D521–6 (Jan 2007), doi: 10.1093/nar/gkl923.
[15] D. S. Wishart, C. Knox, A. C. Guo, et al., HMDB: a knowledgebase for the human metabolome, Nucleic Acids Res 37(Database issue), D603–10 (Jan 2009), doi:10.1093/nar/gkn810.
[16] D. S. Wishart, T. Jewison, A. C. Guo, M. Wilson, C. Knox, et al., HMDB 3.0–The Human Metabolome Database in 2013, Nucleic Acids Res 41(Database issue), D801–7 (Jan 2013), doi:10.1093/nar/gks1065.
[17] J. Griss, A. R. Jones, T. Sachsenberg, M. Walzer, L. Gatto, J. Hartler, G. G. Thallinger, R. M. Salek, C. Steinbeck, N. Neuhauser, J. Cox, S. Neumann, J. Fan, F. Reisinger, Q.-W. Xu, N. Del Toro, Y. Perez-Riverol, F. Ghali, N. Bandeira, I. Xenarios, O. Kohlbacher, J. A. Vizcaino, and H. Hermjakob, The mzTab Data Exchange Format: communicating MS-based proteomics and metabolomics experimental results to a wider audience, Mol Cell Proteomics (Jun 2014), doi: 10.1074/mcp.O113.036681.
[18] S. Böcker, M. C. Letzel, Z. Lipták, and A. Pervukhin, SIRIUS: Decomposing isotope patterns for metabolite identification, Bioinformatics 25(2), 218–224 (2009), doi:10.1093/bioinformatics/btn603.
[19] S. Böcker and K. Dührkop, Fragmentation trees reloaded, J. Cheminform. 8(1), 1–26 (2016), doi:10.1186/s13321-016-0116-8.
[20] K. Dührkop, H. Shen, M. Meusel, J. Rousu, and S. Böcker, Searching molecular structure databases with tandem mass spectra using CSI:FingerID, Proc. Natl. Acad. Sci. 112(41), 12580–12585 (oct 2015), doi:10.1073/pnas.1509788112.
[21] H. L. Röst, G. Rosenberger, P. Navarro, L. Gillet, S. M. Miladinovic, O. T. Schubert, W. Wolski, B. C. Collins, J. Malmstrom, L. Malmström, and R. Aebersold, OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data, Nature Biotechnology 32(3), 219–223 (Mar. 2014).
[22] L. C. Gillet, P. Navarro, S. Tate, H. Röst, N. Selevsek, L. Reiter, R. Bonner, and R. Aebersold, Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis., Molecular & Cellular Proteomics 11(6) (June 2012), doi: 10.1074/mcp.O111.016717.
[23] A. Bertsch, C. Gröpl, K. Reinert, and O. Kohlbacher, OpenMS and TOPP: open source software for LC-MS data analysis., Methods in molecular biology (Clifton, N.J.) 696, 353–367 (2011), doi:10.1007/978-1-60761-987-1_23.
[24] H. L. Röst, T. Sachsenberg, S. Aiche, C. Bielow, H. Weisser, F. Aicheler, S. Andreotti, H.-c. Ehrlich, P. Gutenbrunner, E. Kenar, X. Liang, S. Nahnsen, L. Nilse, J. Pfeuffer, G. Rosenberger, M. Rurik, U. Schmitt, J. Veit, M. Walzer, D. Wojnar, W. E. Wolski, O. Schilling, J. S. Choudhary, L. Malmström, R. Aebersold, K. Reinert, and O. Kohlbacher, OpenMS: a flexible open-source software platform for mass spectrometry data analysis, Nat. Methods 13(9), 741–748 (sep 2016), doi:10.1038/nmeth.3959.
[25] J. Pfeuffer, T. Sachsenberg, O. Alka, M. Walzer, A. Fillbrunn, L. Nilse, O. Schilling, K. Reinert, and O. Kohlbacher, OpenMS - A platform for reproducible analysis of mass spectrometry data, J. Biotechnol. 261(February), 142–148 (2017), doi:10.1016/j.jbiotec.2017.05.016.
[26] L. Reiter, O. Rinner, P. Picotti, R. Huttenhain, M. Beck, M.-Y. Brusniak, M. O. Hengartner, and R. Aebersold, mProphet: automated data processing and statistical validation for large-scale SRM experiments, Nature Methods 8(5), 430–435 (May 2011), doi:10.1038/nmeth.1584.
[27] E. W. Deutsch, M. Chambers, S. Neumann, F. Levander, P.-A. Binz, J. Shofstahl, D. S. Campbell, L. Mendoza, D. Ovelleiro, K. Helsens, L. Martens, R. Aebersold, R. L. Moritz, and M.-Y. Brusniak, TraML—A Standard Format for Exchange of Selected Reaction Monitoring Transition Lists, Molecular & Cellular Proteomics 11(4) (Apr. 2012), doi:10.1074/mcp.R111.015040.
[28] C. Escher, L. Reiter, B. MacLean, R. Ossola, F. Herzog, J. Chilton, M. J. MacCoss, and O. Rinner, Using iRT, a normalized retention time for more targeted measurement of peptides., Proteomics 12(8), 1111–1121 (Apr. 2012), doi:10.1002/ pmic.201100463.
[29] H. L. Röst, G. Rosenberger, P. Navarro, L. Gillet, S. M. Miladinović, O. T. Schubert, W. Wolski, B. C. Collins, J. Malmström, L. Malmström, and R. Aebersold, OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data., Nat. Biotechnol. 32(3), 219–23 (2014), doi:10.1038/nbt.2841.
[30] J. Teleman, H. L. Röst, G. Rosenberger, U. Schmitt, L. Malmström, J. Malmström, and F. Levander, DIANA-algorithmic improvements for analysis of data-independent acquisition MS data, Bioinformatics 31(4), 555–562 (2015), arXiv:9808008, doi:10.1093/bioinformatics/btu686.
[31] K. Dührkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu, and S. Böcker, SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information, Nat. Methods 16(4), 299–302 (apr 2019), doi:10.1038/s41592-019-0344-8.
[32] J. E. Elias and S. P. Gygi, Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry, Nat. Methods 4(3), 207–214 (Mar. 2007).
[33] K. Scheubert, F. Hufsky, D. Petras, M. Wang, L. F. Nothias, K. Dührkop, N. Bandeira, P. C. Dorrestein, and S. Böcker, Significance estimation for large scale metabolomics annotations by spectral matching, Nat. Commun. 8(1) (2017), doi:10.1038/s41467-017-01318-5.