BALL::AmberFF::Option Struct Reference
#include <BALL/MOLMEC/AMBER/amber.h>
List of all members.
Detailed Description
Option names
Definition at line 42 of file amber.h.
Member Data Documentation
automatically assign charges to the system (during setup)
Definition at line 84 of file amber.h.
automatically assign type names to the system (during setup)
Definition at line 88 of file amber.h.
automatically assign types to the system's atoms (during setup)
Definition at line 92 of file amber.h.
use of distance dependent dielectric constant
Definition at line 80 of file amber.h.
Electrostatic cutoff
Definition at line 64 of file amber.h.
Electrostatic cuton
Definition at line 68 of file amber.h.
Nonbonded cutoff. This value is used as cutoff radius in calculations of nonbonded interactions. The unit of this option is 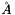 (
(
- See also:
- Default::NONBONDED_CUTOFF)
Definition at line 52 of file amber.h.
during charge assignment, overwrite even non-zero charges
Definition at line 96 of file amber.h.
during charge assignment, overwrite even non-empty type names
Definition at line 100 of file amber.h.
1-4 electrostatic interaction scaling factor.
Definition at line 76 of file amber.h.
1-4 vdw interaction scaling factor.
Definition at line 72 of file amber.h.
Van der Waals cutoff
Definition at line 56 of file amber.h.
Van der Waals cuton
Definition at line 60 of file amber.h.
 1.6.3
1.6.3