|
OpenMS
|
Mass spectrometry (MS) is an essential analytical technique for high-throughput analysis in proteomics and metabolomics. The development of new separation techniques, precise mass analyzers and experimental protocols is a very active field of research. This leads to more complex experimental setups yielding ever increasing amounts of data. Consequently, analysis of the data is currently often the bottleneck for experimental studies. Although software tools for many data analysis tasks are available today, they are often hard to combine with each other or not flexible enough to allow for rapid prototyping of a new analysis workflow.
OpenMS, a software framework for rapid application and method development in mass spectrometry has been designed to be portable, easy-to-use, and robust while offering a rich functionality ranging from basic data structures to sophisticated algorithms for data analysis (https://www.nature.com/nmeth/journal/v13/n9/abs/nmeth.3959.html).
Ease of use: OpenMS follows the object-oriented programming paradigm, which aims at mapping real-world entities to comprehensible data structures and interfaces. OpenMS enforces a coding style that ensures consistent names of classes, methods and member variables which increases the usability as a software library. Another important feature of a software framework is documentation. We decided to use doxygen (www.doxygen.org/) to generate the class documentation from the source code, which ensures consistency of code and documentation. The documentation is generated in HTML format making it easy to read with a web browser.
Robustness: Robustness of algorithms is essential if a new method will be applied routinely to large scale datasets. Typically, there is a trade-off between performance and robustness. OpenMS tries to address both issues equally. In general, we try to tolerate recoverable errors, e.g. files that do not entirely fulfill the format specifications. On the other hand, exceptions are used to handle fatal errors. To check for correctness, more than 1000 unit tests are implemented in total, covering public methods of classes. These tests check the behavior for both valid and invalid use. Additionally, preprocessor macros are used to enable additional consistency checks in debug mode, enforce pre- and post-conditions, and are then disabled in productive mode for performance reasons.
Extensibility: Since OpenMS is based on several external libraries it is designed for the integration of external code. All classes are encapsulated in the OpenMS namespace to avoid symbol clashes with other libraries. Through the use of C++ templates, many data structures are adaptable to specific use cases. Also, OpenMS supports standard formats and is itself open-source software. The use of standard formats ensures that applications developed with OpenMS can be easily integrated into existing analysis pipelines. OpenMS source code is released under the permissive BSD 3 license and located on GitHub, a repository for open-source software. This allows users to participate in the project and to contribute to the code base.
Scriptable: OpenMS allows exposing its functionality through python bindings (pyOpenMS). This eases the rapid development of algorithms in Python that later can be translated to C++. Please see our pyOpenMS documentation for a description and walk-through of the pyOpenMS capabilities.
Portability: OpenMS supports Windows, Linux, and OS X platforms.
The following image shows the overall structure of OpenMS:
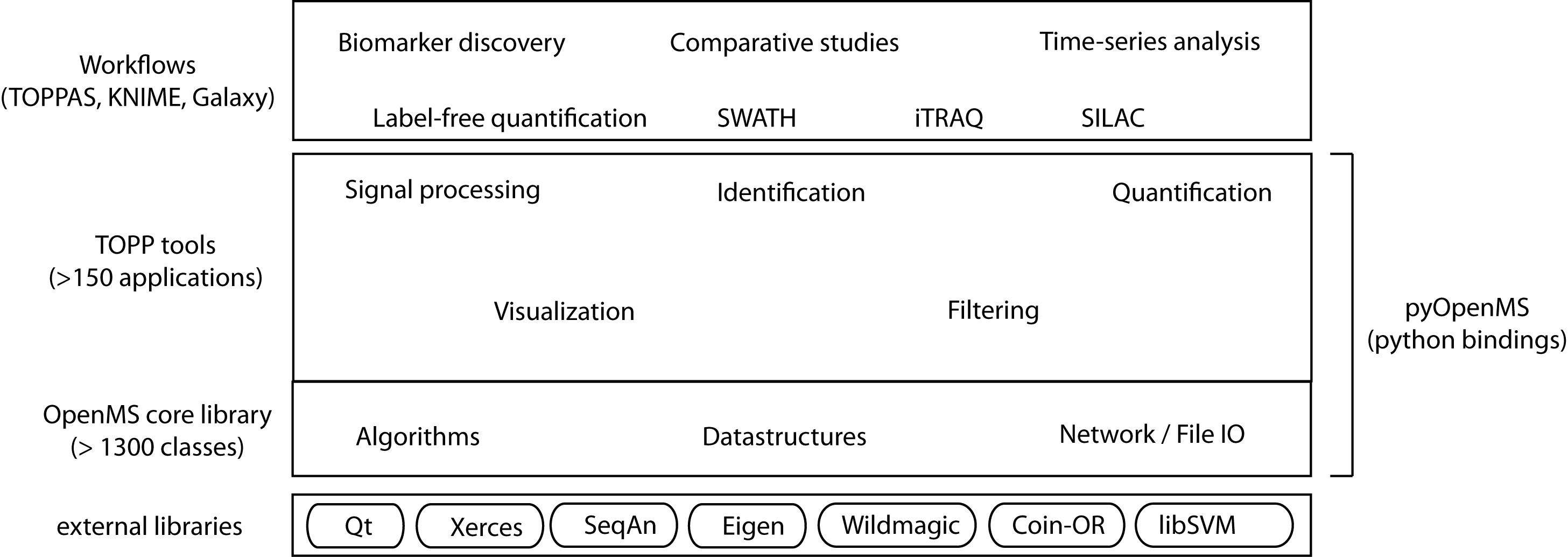
The structure of the OpenMS framework.
The OpenMS software framework consists of three main layers:
Each level of increasing abstraction provides better usability, but limits the extensibility as the Python and workflow levels only have access to the exposed Python API or the available set of TOPP tools respectively. Increasing abstraction, however, makes it easier to design and execute complex analyses, even across multiple omics types. By following a layered design the different needs of bioinformaticians and life scientists are addressed.
Before we get started developing with OpenMS, we would like to point to some information on the development model and conventions we follow to maintain a coherent code base.
Development model
OpenMS follows the Gitflow development workflow which is excellently describedI here. Additionally we encourage every developer (even if he is eligible to push directly to OpenMS) to create his own fork (e.g. username). The GitHub people provide superb documentation on forking and how to keep your fork up-to-date. With your own fork you can follow the Gitflow development model directly, but instead of merging into "develop" in your own fork you can open a pull request. Before opening the pull request, please check the checklist.
Some more details and tips are collected here.
Conventions
See the manual for proper coding style: Coding conventions also see: C++ Guide. We automatically test for common coding convention violations using a modified version of cpplint. Style testing can be enabled using CMake options. We also provide a configuration file for Uncrustify for automated style corrections (see "tools/uncrustify.cfg").
Commit Messages
In order to ease the creation of a CHANGELOG we use a defined format for our commit messages. See the manual for proper commit messages: How to write commit messages.
Automated Unit Tests
Pull requests are automatically tested using our continuous integration platform. In addition we perform nightly test runs covering different platforms. Even if everything compiled well on your machine and all tests passed, please check if you broke another platform on the next day. Nightly tests: CDASH
Experimental Installers
We automatically build installers for different platforms. These usually contain unstable or partially untested code - so use them at your own risk. The nightly (unstable) installers are available here.
Technical Documentation
Documentation of classes and tools is automatically generated using doxygen: See the documentation for HEAD See the documentation for the latest release branch
Building OpenMS
Before you get started coding with OpenMS you need to build it for your operating system. Please follow the build instructions from the documentation.
Building OpenMS on GNU/Linux
Building OpenMS on Mac OS X
Building OpenMS on Windows
Note that for development purposes, you might want to set the variable CMAKE_BUILD_TYPE to Debug. Otherwise, the default Release will be applied and disables pre-condition and post-condition checks, and assertions.
Choice of an IDE
You are, of course, free to choose your favorite (or even no) IDE for OpenMS development but given the size of OpenMS, not all IDEs perform equally well. We have good experiences with Qt Creator on Linux and Mac, because it can directly import CMake Projects and is rather fast in indexing all files. On Windows, Visual Studio is currently the preferred solution. Additionally, you may want to try JetBrains CLion (it is free for students, teachers and open source projects). Another option is Eclipse with C++ support, which can also import CMake projects directly with the respective CMake generator.
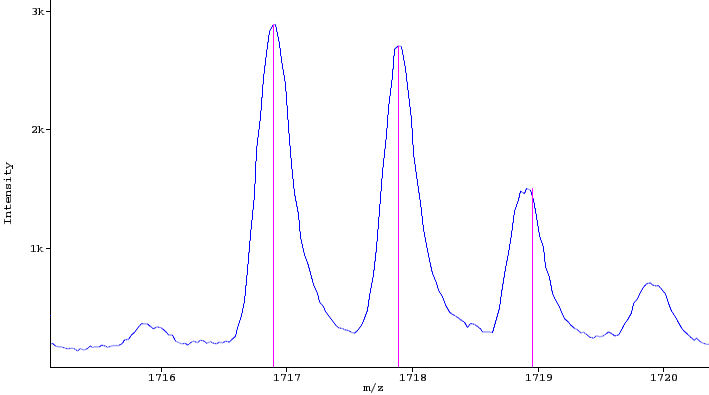
The following terms for MS-related data are used in this tutorial and the OpenMS class documentation:
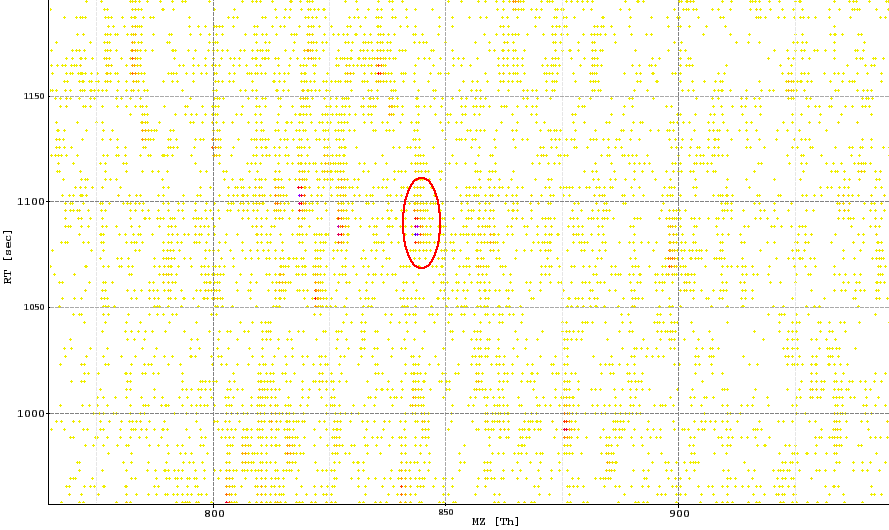
The image below shows a peak map and the red circle highlights a feature.
The extensible OpenMS library implements common mass spectrometric data processing tasks through a well defined API in C++ and Python using standardized open data formats.
OpenMS provides algorithms in many fields of computational metabolomics and proteomics.
The following list is intended to algorithm and tool developers a starting point to tools and classes relevant to their scientific question at hand. It does not include third-party tools but only tools that were implemented in OpenMS.
| Folder | Description |
|---|---|
| openms | Source code of core library |
| openms_gui | Source code of GUI applications (e.g.: TOPPView) |
| topp | Source code of (stable) OpenMS Tools |
| util | Source code of (experimental) OpenMS Tools |
| pyOpenMS | Source files providing the python bindings |
| tests | Source code of class and tool tests |
| Folder | Description |
|---|---|
| ANALYSIS | Source code of high-level analysis like PeakPicking, Quantitation, Identification, MapAlignment |
| APPLICATIONS | Source code for tool base and handling |
| CHEMISTRY | Source code dealing with Elements, Enzymes, Residues/Amino Acids, Modifications, Isotope distributions and amino acid sequences |
| COMPARISON | Different scoring functions for clustering and spectra comparison |
| CONCEPT | OpenMS concepts (types, macros, ...) |
| DATASTRUCTURES | Auxiliary data structures |
| FILTERING | Filter |
| FORMAT | Source code for I/O classes and file formats |
| INTERFACES | Interfaces (WIP) |
| KERNEL | Core data structures |
| MATH | Source code for math functions and classes |
| METADATA | Source code for classes that capture metadata about a MS or HPLC-MS experiment |
| SIMULATION | Source code of MS simulator |
| SYSTEM | Source code for basic functionality (file system, stopwatch) |
| TRANSFORMATIONS | Feature detection (MS1 label-free and isotopic labelling) and PeakPickers (centroiding algorithms) |
Within the ANALYSIS folder, you can find several important tools
| Folder | Description |
|---|---|
| DECHARGING | Algorithms for de-charging (charge analysis) for peptides and metabolites |
| DENOVO | Algorithms for "de-novo" identification tools including CompNovo |
| ID | Source code dealing with identifications including ID conflict resolvers, metabolite spectrum matching and target-decoy models |
| MAPMATCHING | Algorithms for retention time correction and feature matching (matching between runs) |
| MRM | Algorithms for MRM Fragment selection |
| OPENSWATH | OpenSWATH algorithms for targeted, chromatogram-based analysis of MRM, SRM, PRM, DIA and SWATH-MS data |
| PIP | Peak intensity predictor |
| QUANTITATION | Algorithms for quantitative analysis including isobaric labelling |
| RNPXL | Algorithms for RNA cross-linking |
| SVM | Algorithms for SVM |
| TARGETED | Algorithms for targeted proteomics (MRM, SRM) |
| XLMS | Algorithms for Cross-link mass spectrometry |
For the sake of completeness you will find a short list of the THIRDPARTY tools, which are integrated via wrappers into the OpenMS framework (usually called -Adapter e.g. SiriusAdapter)
Wrapper to third-party tools:
The OpenMS kernel contains the data structures that store the actual MS data.
For storing the basic MS data (spectra, chromatograms, and full runs) OpenMS uses
For storing quantified peptides or analytes in single MS runs, OpenMS uses so called feature maps.
The main data structures for quantitative information are
To store quantified peptides or analytes over several MS runs, OpenMS uses so called consensus maps.
To store identified peptides OpenMS has classes
| Stored Entity | Class Name |
|---|---|
| Mass Peak (m/z + intensity) | Peak1D |
| Elution Peak (rt + intensity) | ChromatogramPeak |
| Spectrum of Mass Peaks | MSSpectrum |
| Chromatogram of Elution Peaks | MSChromatogram |
| Mass trace for small molecule detection | MassTrace |
| Full MS run, containing both spectra and chromatograms | MSExperiment (alias PeakMap) |
| Feature (isotopic pattern of eluting analyte) | Feature |
| All features detected in an MS Run | FeatureMap |
| Linked / Grouped feature (e.g., same Peptide quantified in several MS runs) | ConsensusFeature |
| All grouped ConsensusFeatures of a multi-run experiment | ConsensusMap |
| Peptide Spectrum Match | PeptideHit |
| Identified Spectrum with one or several PSMs | PeptideIdentification |
| Identified Protein | ProteinHit |
OpenMS provides one-, two- and d-dimensional data points, either with or without metadata attached to them.
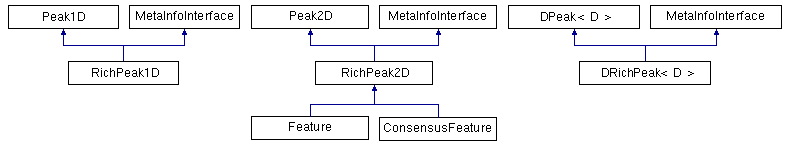
One-dimensional data points: One-dimensional data points (Peak1D) are the most important ones and used throughout OpenMS. The two-dimensional and d-dimensional data points are needed rarely and used for special purposes only. Peak1D provides getter and setter methods to store the mass-to-charge ratio and intensity.
Two-dimensional data points: The two-dimensional data points store mass-to-charge, retention time and intensity. The most prominent example we will later take a closer look at is the Feature class, which stores a two-dimensional position (m/z and RT) and intensity of the eluting peptide or analyte.
The base class of the two-dimensional data points is Peak2D. It provides the same interface as Peak1D and additional getter and setter methods for the retention time. RichPeak2D is derived from Peak2D and adds an interface for metadata. The Feature is derived from RichPeak2D and adds information about the convex hull of the feature, quality and so on.
For information on d-dimensional data points see the appendix.
The most important container for raw/profile data and centroided peaks is MSSpectrum. The elements of a MSSpectrum are peaks (Peak1D). In fact it is so common that it has its own typedef PeakSpectrum. MSSpectrum is derived from SpectrumSettings, a container for the metadata of a spectrum (e.g. precursor information). Here, only MS data handling is explained, SpectrumSettings is described in subsection meta data of a spectrum. In the following example (Tutorial_MSSpectrum.cpp) program, a MSSpectrum is filled with peaks, sorted according to mass-to-charge ratio and a selection of peak positions is displayed as One-dimensional data points:
Example: Tutorial_MSSpectrum.cpp
In this example, we create MS1 spectrum at 1 minute and insert peaks with descending mass-to-charge ratios (for educational reasons). We sort the peaks according to ascending mass-to-charge ratio. Finally we print the peak positions of those peaks between 800 and 1000 Thomson. For printing all the peaks in the spectrum, we simply would have used the STL-conform methods begin() and end(). In addition to the iterator access, we can also directly access the peaks via vector indices (e.g. spectrum[0] is the first Peak1D object of the MSSpectrum).
The most important container for targeted analysis / XIC data is MSChromatogram. The elements of a MSChromatogram are chromatogram peaks (Peak1D). MSChromatogram is derived from ChromatogramSettings, a container for the metadata of a chromatogram (e.g. containing precursor and product information), similarly to SpectrumSettings. In the following example (Tutorial_MSChromatogram.cpp) program, a MSChromatogram is filled with chromatographic peaks, sorted according to retention time and a selection of peak positions is displayed.
Example: Tutorial_MSChromatogram
Fill MSChromatogram with chromatographic peaks, sorted according to retention time
Since much of the functionality is shared between MSChromatogram and MSSpectrum, further examples can be gathered from the MSSpectrum subsection.
The precursor data stored along with MS/MS spectra contains invaluable information for MS/MS analysis (e.g, m/z, charge, activation mode, collision energy). This information is stored in Precursor objects that can be retrieved from each spectrum. For a complete list of functions please see the Precursor class documentation.
Example: Tutorial_Precursor
Retrieve precursor information
The targeted analysis of SRM or DIA (SWATH-MS) type of data requires a set of targeted assays as well as raw data chromatograms. The MRMTransitionGroup class allows users to map these two types of information and store them together with identified features conveniently in a single object.
Example: Tutorial_MRMTransitionGroup
Create an empty MRMTransitionGroup with two dummy transitions
Note how the identifiers of the chromatograms and the assay information (ReactionMonitoringTransition) are matched so that downstream algorithms can utilize the meta-information stored in the assays for data analysis.
Although raw data maps, peak maps and feature maps are conceptually very similar they are stored in different data types. For raw data and peak maps, the default container is MSExperiment, which is an array of MSSpectrum instances. In contrast to raw data and peak maps, feature maps are not a collection of one-dimensional spectra, but an array of two-dimensional feature instances. The main data structure for feature maps is called FeatureMap.
Although MSExperiment and FeatureMap differ in the data they store, they also have things in common. Both store metadata that is valid for the whole map, i.e. sample description and instrument description. This data is stored in the common base class ExperimentalSettings.
MSExperiment contains ExperimentalSettings (metadata of the MS run) and a vector<MSSpectrum>. The one-dimensional spectrum MSSpectrum is derived from SpectrumSettings (metadata of a spectrum).
Example: Tutorial_MSExperiment.cpp
The following example creates a MSExperiment containing four MSSpectrum instances. We then iterate over RT range (2,3) and m/z range (603,802) and print the peak positions using an AreaIterator. Then we show how we iterate over all spectra and peaks. In the commented out part, we show how to load/store all spectra and associated metadata from/to an mzML file.
FeatureMap, the container for features, is simply a vector<Feature>. Additionally, it is derived from ExperimentalSettings, to store the meta information. All peak and feature containers (MSSpectrum, MSExperiment, FeatureMap) are also derived from RangeManager. This class facilitates the handling of MS data ranges. It allows to calculate and store both the position range and the intensity range of the container.
Example: Tutorial_FeatureMap.cpp
The following examples creates a FeatureMap containing two Feature instances. Then we iterate over all features and output the retention time and m/z. We then show, how to use the underlying range manager to retrieve FeatureMap boundaries in rt, m/z, and intensity.
| mzML | The HUPO-PSI standard format for mass spectrometry data |
| mzIdentML | The HUPO-PSI standard format for identification results data from any search engines |
| mzTAB | The HUPO-PSI standard format for reporting MS-based proteomics and metabolomics results |
| traML | The HUPO-PSI standard format for exchange and transmission lists for selected reaction monitoring (SRM) experiments |
| featureXML | The OpenMS format for quantitation results |
| consensusXML | The OpenMS format for grouping features in one map or across several maps |
| idXML | The OpenMS format for identification results |
| trafoXML | The OpenMS format for storing of transformations |
| OpenSWATH |
For further information of the HUPO Proteomics Standards Initiative please visit: http://www.psidev.info/
To make direct output to std::out and std::err more consistent, OpenMS provides several low-level macros:
OPENMS_LOG_FATAL_ERROR,
OPENMS_LOG_ERROR
OPENMS_LOG_WARN,
OPENMS_LOG_INFO and
OPENMS_LOG_DEBUG
which should be used instead of the less descriptive std::out and std::err streams.
If you are writing an OpenMS tool, you can also use the ProgressLogger to indicate how many percent of the processing has already been performed:
Example: Tutorial_Logger.cpp
Logging the Tool Progress
Depending on how the user configures the tool, this output is written to the command line or a log file.
Identifications of proteins, peptides, and the mapping between peptides and proteins (or groups of proteins) are stored in dedicated data structures. These data structures are typically stored to disc as idXML or mzIdentML file. The highest-level structure is ProteinIdentification. It stores all identified proteins of an identification run as ProteinHit objects + additional metadata (search parameters, etc.). Each ProteinHit contains the actual protein accession, an associated score, and (optionally) the protein sequence. A ProteinIdentification object stores the data corresponding to a single identified spectrum or feature. It has members for the retention time, m/z, and a vector of PeptideHits. Each PeptideHit stores the information of a specific peptide-to-spectrum match (e.g., the score and the peptide sequence). Each PeptideHit also contains a vector of PeptideEvidence objects which store the reference to one (or in the case the peptide maps to multiple proteins multiple) Proteins and the position therein.
Example: Tutorial_IdentificationClasses.cpp
Create all identification data needed to store an idXML file
An Element object is the representation of an element. It can store the name, symbol and mass (average/mono) and natural abundances of isotopes. Elements are retrieved from the ElementDB singleton. The EmpiricalFormula object can be used to represent the empirical formula of a compound as well as to extract its natural isotope abundance and weight.
Example: Tutorial_Element.cpp
Work with Element object
Example: Tutorial_EmpiricalFormula.cpp
Extract isotope distribution and monoisotopic weight of an EmpiricalFormula object
An AASequence object stores a (potentially chemically modified) peptide. It can conveniently be constructed from the amino acid sequence (e.g., a string or a string literal “DEFIANGR”). Modifications may be encoded using the unimod name. Once constructed, many convenient functions are available to calculate peptide or ion properties.
Example: Tutorial_AASequence.cpp
Compute and output basic AASequence properties
Internally, an AASequence object is composed of Residues.
Residues are the building blocks of AASequence objects. They store physico-chemical properties of amino acids. ResidueDB provides access to different residue sets (e.g. all natural AAs).
Example: Tutorial_Residue.cpp
Compute and output basic Residue properties
If a residue is modified (e.g. phosphorylation of an amino acid) it can be stored in the ResidueModification class. The ResidueModification class stores information about chemical modifications of residues. Each ResidueModification has an ID, the residue that can be modified with this modification and the difference in mass between the unmodified and the modified residue, among other information. The Residue class allows to set one modification per residue and the mass difference of the modification is accounted for in the mass of the residue. The class ModificationsDB is a database of ResidueModifications. These are mostly initialized from the file “/share/CHEMISTRY/unimod.xml” containing a slightly modified version of the UniMod database of modifications. ModificationsDB has functions to search for modifications by name or mass.
Example: Tutorial_ResidueModification.cpp
Set a ResidueModification on a Residue
The TheoreticalSpectrumGenerator generates ion ladders from AASequences.
Example: Tutorial_TheoreticalSpectrumGenerator.cpp
Generate theoretical spectra
OpenMS provides the most common digestion enzymes (DigestionEnzymeProtein) used in MS. They are stored in the ProteaseDB singleton and loaded from “/share/CHEMISTRY/Enzymes.xml”.
Example: Tutorial_Enzyme.cpp
Digest amino acid sequence
TOPP (The OpenMS Pipeline) tools are small command line applications built using the OpenMS library. They act as building blocks for complex analysis workflows and may perform e.g. simple signal processing tasks like filtering, up to more complex tasks like protein inference and quantitation over several MS runs. Common to all TOPP tools is a command line interface allowing automatic integration into workflow engines like KNIME. They are the preferred way to integrate novel methods as application into OpenMS. When we first create a novel TOPP tool it is considered unstable. To set it apart from the stable and well tested tools it gets first created as TOPP Util (note: the name “util” has historic reasons and may be changed to unstable tools in the future). If it is well tested it will be promoted to a stable Tool in future OpenMS versions.
Imagine that you want to create a new tool that allows filtering of sequence databases. What you usually would first do is check if such or similar functionality has already been implemented in any of the >150 TOPP tools. If you are unsure which one to use, just ask on the mailing list, the gitter chat or contact one of the developers directly. The following subsection demonstrates how the original “DatabaseFilter” tool was created from scratch an integrated into OpenMS. Basically any tool you want to integrate needs to follow the steps outlined below.
But let’s first get started by defining what our tool should actually do: The DatabaseFilter tool should provide functionality to reduce a fasta database by filtering its entries based on different criteria. A simple criterion could be the length of a protein. To make the task a bit more interesting and to show other parts of the OpenMS library, we will start with a bit more complex filtering step that keeps all entries from the fasta database that have been identified in a peptide search (e.g., using X!Tandem, Mascot or MSGF+). This functionality might come in handy if the size of large databases needs to be reduced to a manageable size. In addition, we want the user to be able to choose between keeping and removing matching protein id.
Example: Tutorial_Template.cpp
Template for OpenMS tool development
Define tool parameters Each TOPP tool defines a set of parameters that will be available from the command line, KNIME, and other workflow systems. This is done in the void registerOptionsAndFlags_() method. In our case we want to read a protein database (fasta format), a file containing identification data (idXML format), and an option to switch between keeping (whitelisting) and removing (blacklisting) entries based on the filter result. This is our input. The reduced database forms the output and should be written to a protein database in fasta format. This is easily done by adding following lines to:
Example: Tutorial_Final.cpp
Registration of tool parameters
Functions, classes and references can be checked in the OpenMS / TOPP documentation (ftp://ftp.mi.fu-berlin.de/pub/OpenMS/release-documentation/html/index.html)
After a tool is executed, the registered parameters are available in the main_ function of the TOPP tool and can be read using the getStringOption_ method. Special methods for integers, lists and floating point parameters exist and are in the TOPPBase documentation but are not needed for this example.
Example: Tutorial_Final.cpp
First the different file formats and data structures for peptide identifications have to be included at the top of the file.
Example: Tutorial_Final.cpp
Add essential includes
Read the input files
Note: both peptide_identifications and protein_identifications contain protein accessions. The difference between them is that protein_identifications only contain the inferred set of protein accessions while peptide_identifications contains all protein accessions the peptides map to. We consider only the larger set of protein accessions stored in the peptide identifications. In principle, it would be easy to add another parameter that adds a filter for the inferred accessions stored in protein_identifications.
First, the accessions are extracted from the IdXML file. Here knowledge of the data structure is needed to extract the protein accessions. The class PeptideIdentification stores general information about a single identified spectrum (e.g., retention time, precursor mass-to-charge). A vector of PeptideHits is stored in each PeptideIdentification object and represent the potentially multiple PSMs of a single spectrum. They can be returned by calling .getHits(). Each peptide sequence stored in a PeptideHit may map to one or multiple proteins. This peptide to protein mapping information is stored in a vector of PeptideEvidence accessible by .getPeptideEvidences(). From each of these evidences we can extract the protein accession with .getProteinAccession().
To store all proteins accessions in the set id_accessions, we write:
Example: Tutorial_Final.cpp
Store protein accessions
Now that we assembled the set of all protein accessions we are ready to compare them to the fasta_accessions. If they are similar and the method whitelist or they are different and the method blacklist was chosen, the fasta entries are copied to the new fasta database.
Example: Tutorial_Final.cpp
Add method functionality
Example: Tutorial_Final.cpp
Write the output
Testing your tools is essential and required to promote your experimental util to an official TOPP tool. It is not mandatory to provide a test for a util but appreciated. For this test a .fasta and a compatible .idXML file have to be added to /src/tests/topp/. Further the test procedure has to be added to CMakeLists.txt in the same folder.
Example: Tutorial_Test.cpp
Add tests
These tests run the program with the given parameters and then call a diff tool to compare the generated output to the expected output.
We add it to the UTILS docu page (in doc/doxygen/public/UTILS.doxygen). Later (when we have a working application) we will write an application test (this is optional but recommended for Utils. For Tools it is mandatory). See TOPP tools above and add the test to the bottom of src/tests/topp/CMakeLists.txt.
This is how a util should look after code polishing: Here, the support for different formats was extended (idXML and MZIdentML). Since different filter criteria may be introduced in the future, the structure was slightly changed with a function for the filtering by ID (filterByProteinIDs_) - in order to allow higher flexibility when adding new a functionality later on.
Example: Tutorial_final.cpp
Polish your code - add additional functionality
Afterwards you can commit your changes to a new branch “feature/DatabaseFilter” of your OpenMS clone on github and submit a pull request on your github page. After a short review process by the OpenMS Team, the tool will be added the OpenMS Library.
The d-dimensional data points are needed in special cases only, e.g. in template classes that operate in any number of dimensions. The base class of the d-dimensional data points is DPeak. The methods to access the position are getPosition and setPosition. Note that the one-dimensional and two-dimensional data points also have the methods getPosition and setPosition. They are needed in order to be able to write algorithms that can operate on all data point types. It is, however, recommended not to use these members unless you really write such a generic algorithm.
If OpenMS TOPP_tools and UTILS_tools are not sufficient for a certain scenario, you can either request changes to OpenMS or modify/extend your own fork of OpenMS. A third alternative is using OpenMS as a dependency while not touching OpenMS itself. Once you've finished your new tool, and it runs on the development machine, you're done. If you want to develop with OpenMS as external project have a look the example code ( /share/OpenMS/examples/external_code/).
 1.9.4
1.9.4 
