| ►C_Alloc_base< _Tp, _Alloc > | |
| CKDTree< 2, KDTreeFeatureNode > | |
| ►C_Alloc_base< _Val, std::allocator< _Node< _Val > > > | |
| CKDTree< __K, _Val, _Acc, _Dist, _Cmp, _Alloc > | |
| ►C_Base_iterator | |
| C_Iterator< _Val, _Ref, _Ptr > | |
| C_Bracket_accessor< _Val > | |
| ►C_Node_base | |
| C_Node< _Val > | |
| C_Node_compare< _Val, _Acc, _Cmp > | |
| C_Region< __K, _Val, _SubVal, _Acc, _Cmp > | |
| CAAcid_ | |
| CAAIndex | Representation of selected AAIndex properties |
| CAASequence | Representation of a peptide/protein sequence |
| COPXLDataStructs::AASeqWithMass | The AASeqWithMass struct represents a normal peptide with its precomputed mass |
| COPXLDataStructs::AASeqWithMassComparator | The AASeqWithMassComparator is a comparator for AASeqWithMass objects |
| CFuzzyStringComparator::AbortComparison | Internal exception class |
| CAbsoluteQuantitationMethod | AbsoluteQuantitationMethod is a class to hold information about the quantitation method and for applying and/or generating the quantitation method |
| CAbsoluteQuantitationStandards | AbsoluteQuantitationStandards is a class to handle the relationship between runs, components, and their actual concentrations |
| CAbsoluteQuantitationStandardsFile | Load files containing runConcentration data |
| CSiriusMSFile::AccessionInfo | < class to store information about accessions |
| CAccurateMassSearchResult | |
| CAcqusHandler | Read-only acqus File handler for XMass Analysis |
| CTOPPViewMenu::ActionRequirement_ | |
| CAdduct | |
| CAdductInfo | |
| CMSstatsFile::AggregatedConsensusInfo | |
| CTriqlerFile::AggregatedConsensusInfo | |
| CAhoCorasickAmbiguous | Extended Aho-Corasick algorithm capable of matching ambiguous amino acids in the pattern (i.e. proteins) |
| Calways_true< _Tp > | |
| CPepXMLFile::AminoAcidModification | |
| CMzIdentMLDOMHandler::AnalysisSoftware | Struct to hold the used analysis software for that file |
| CMSQuantifications::AnalysisSummary | |
| CNucleicAcidSearchEngine::AnnotatedHit | |
| CSimpleSearchEngineAlgorithm::AnnotatedHit_ | Slimmer structure as storing all scored candidates in PeptideHit objects takes too much space |
| ►CAnnotation1DItem | An abstract class acting as an interface for the different 1D annotation items |
| CAnnotation1DCaret | An annotation item which paints a set of carets on the canvas |
| CAnnotation1DDistanceItem | An annotation item which represents a measured distance between two peaks |
| CAnnotation1DPeakItem | A peak annotation item |
| CAnnotation1DTextItem | An annotation item which represents an arbitrary text on the canvas |
| CAnnotation1DVerticalLineItem | An annotation item which represents a vertical line and text label on top |
| CAnnotationStatistics | |
| CMassTraceDetection::Apex | |
| CAppliedProcessingStep | A processing step that was applied to a data item, possibly with associated scores |
| CMSQuantifications::Assay | |
| CQcMLFile::Attachment | Representation of an attachment |
| CAveragePosition< D > | Maintain an average position by summing up positions with weights |
| CAxisPainter | Draws a coordinate axis. It has only static methods, that's why the constructor is private |
| CAxisTickCalculator | Calculates ticks for a given value range |
| CBase64 | Class to encode and decode Base64 |
| CBaseVisualizer< ObjectType > | A base class for all visualizer classes |
| ►CBaseVisualizer< Acquisition > | |
| CAcquisitionVisualizer | Class that displays all meta information for Acquisition objects |
| ►CBaseVisualizer< AcquisitionInfo > | |
| CAcquisitionInfoVisualizer | Class that displays all meta information for AcquisitionInfo objects |
| ►CBaseVisualizer< ContactPerson > | |
| CContactPersonVisualizer | Class that displays all meta information for ContactPerson objects |
| ►CBaseVisualizer< DataProcessing > | |
| CDataProcessingVisualizer | Class that displays all meta information for DataProcessing objects |
| ►CBaseVisualizer< Digestion > | |
| CDigestionVisualizer | Class that displays all meta information of digestion objects |
| ►CBaseVisualizer< DocumentIdentifier > | |
| CDocumentIdentifierVisualizer | Class that displays all meta information for DocumentIdentifier objects |
| ►CBaseVisualizer< ExperimentalSettings > | |
| CExperimentalSettingsVisualizer | Class that displays all meta information for ExperimentalSettings objects |
| ►CBaseVisualizer< Gradient > | |
| CGradientVisualizer | GradientVisualizer is a visualizer class for objects of type gradient |
| ►CBaseVisualizer< HPLC > | |
| CHPLCVisualizer | Class that displays all meta information for HPLC objects |
| ►CBaseVisualizer< Instrument > | |
| CInstrumentVisualizer | Class that displays all meta information for an MS instrument |
| ►CBaseVisualizer< InstrumentSettings > | |
| CInstrumentSettingsVisualizer | Class that displays all meta information for InstrumentSettings objects |
| ►CBaseVisualizer< IonDetector > | |
| CIonDetectorVisualizer | Class that displays all meta information for IonDetector objects |
| ►CBaseVisualizer< IonSource > | |
| CIonSourceVisualizer | Class that displays all meta information for IonSource objects |
| ►CBaseVisualizer< MassAnalyzer > | |
| CMassAnalyzerVisualizer | Class that displays all meta information for MassAnalyzer objects |
| ►CBaseVisualizer< MetaInfoDescription > | |
| CMetaInfoDescriptionVisualizer | Class that displays all meta information for MetaInfoDescription objects |
| ►CBaseVisualizer< MetaInfoInterface > | |
| CMetaInfoVisualizer | MetaInfoVisualizer is a visualizer class for all classes that use one MetaInfo object as member |
| ►CBaseVisualizer< Modification > | |
| CModificationVisualizer | Class that displays all meta information of modification objects |
| ►CBaseVisualizer< PeptideHit > | |
| CPeptideHitVisualizer | Class that displays all meta information for PeptideHit objects |
| ►CBaseVisualizer< PeptideIdentification > | |
| CPeptideIdentificationVisualizer | Class that displays all meta information for PeptideIdentification objects |
| ►CBaseVisualizer< Precursor > | |
| CPrecursorVisualizer | Class that displays all meta information for Precursor objects |
| ►CBaseVisualizer< Product > | |
| CProductVisualizer | Class that displays all meta information for Product objects |
| ►CBaseVisualizer< ProteinHit > | |
| CProteinHitVisualizer | Class that displays all meta information for ProteinHit objects |
| ►CBaseVisualizer< ProteinIdentification > | |
| CProteinIdentificationVisualizer | Class that displays all meta information for ProteinIdentification objects |
| ►CBaseVisualizer< Sample > | |
| CSampleVisualizer | Class that displays all meta information of sample objects |
| ►CBaseVisualizer< ScanWindow > | |
| CScanWindowVisualizer | Class that displays all meta information for ScanWindow objects |
| ►CBaseVisualizer< Software > | |
| CSoftwareVisualizer | Class that displays all meta information for Software objects |
| ►CBaseVisualizer< SourceFile > | |
| CSourceFileVisualizer | Class that displays all meta information for SourceFile objects |
| ►CBaseVisualizer< SpectrumSettings > | |
| CSpectrumSettingsVisualizer | Class that displays all meta information for SpectrumSettings objects |
| ►CBaseVisualizer< Tagging > | |
| CTaggingVisualizer | Class that displays all meta information of tagging objects |
| ►Cbasic_string< Char > | STL class |
| ►Cstring | STL class |
| CString | A more convenient string class |
| CBasicStatistics< RealT > | Calculates some basic statistical parameters of a distribution: sum, mean, variance, and provides the normal approximation |
| CBasicStatistics< CoordinateType > | |
| ►CBasicStatistics< double > | |
| CAsymmetricStatistics< RealT > | Internal class for asymmetric distributions |
| CBilinearInterpolation< Key, Value > | Provides access to bilinearly interpolated values (and derivatives) from discrete data points. Values beyond the given range of data points are implicitly taken as zero |
| CMzMLHandlerHelper::BinaryData | Representation for binary data in mzML |
| CBinaryDataArray | The datastructures used by the OpenSwath interfaces |
| CBinaryTreeNode | Elements of a binary tree used to represent a hierarchical clustering process |
| CEnzymaticDigestionLogModel::BindingSite_ | |
| ►CBinInputStream | |
| CBzip2InputStream | Implements the BinInputStream class of the xerces-c library in order to read bzip2 compressed XML files |
| CGzipInputStream | Implements the BinInputStream class of the xerces-c library in order to read gzip compressed XML files |
| CBinnedSpectrum | This is a binned representation of a PeakSpectrum |
| CBitsPerValue< AAcid > | |
| CFeatureFinderAlgorithmIsotopeWavelet::BoxElement | Internally used data structure for the sweep line algorithm |
| CIsotopeWaveletTransform< PeakType >::BoxElement | Internally used data structure |
| CBSpline< T > | |
| CBSpline2d | B spline interpolation |
| CBSpline< double > | |
| CQTCluster::BulkData | Class to store the bulk internal data (neighbors, annotations, etc.) |
| CBzip2Ifstream | Decompresses files which are compressed in the bzip2 format (*.bz2) |
| ►CCachedmzML | An class that uses on-disk caching to read and write spectra and chromatograms |
| CSpectrumAccessOpenMSCached | An implementation of the Spectrum Access interface using on-disk caching |
| CInternalCalibration::CalibrantStats_ | Statistics when adding peptide calibrants |
| CCalibrationData | A helper class, holding all calibration points |
| CItraqConstants::ChannelInfo | Stores information on an iTRAQ channel |
| CChargePair | Representation of a (putative) link between two Features, which stem from the same compound but have different charge (including different adduct ions (H+, Na+, ..) |
| CChromatogram | A single chromatogram |
| CMzMLHandler::ChromatogramData | Data necessary to generate a single chromatogram |
| CChromatogramMeta | Identifying information for a chromatogram |
| CChromatogramPeak | A 1-dimensional raw data point or peak for chromatograms |
| CChromatogramTools | Conversion class to convert chromatograms |
| CChromeleonFile | Load Chromeleon HPLC text file and save it into a `MSExperiment` |
| CChromExtractParams | ChromatogramExtractor parameters |
| CMSSpectrum::Chunk | Used to remember what subsets in a spectrum are sorted already to allow faster sorting of the spectrum |
| CMSSpectrum::Chunks | |
| CCitation | Stores Citations for individual TOPP tools |
| CEnzymaticDigestionLogModel::CleavageModel_ | |
| COPXLDataStructs::CLSMScoreComparator | Comparator to sort CrossLinkSpectrumMatches by the main score |
| CClusterAnalyzer | Bundles analyzing tools for a clustering (given as sequence of BinaryTreeNode's) |
| ►CClusterFunctor | Base class for cluster functors |
| CAverageLinkage | AverageLinkage ClusterMethod |
| CCompleteLinkage | CompleteLinkage ClusterMethod |
| CSingleLinkage | SingleLinkage ClusterMethod |
| CClusterHierarchical | Hierarchical clustering with generic clustering functions |
| CClusteringGrid | Data structure to store 2D data to be clustered e.g. (m/z, retention time) coordinates from multiplex filtering |
| CClusterProxyKD | Proxy for a (potential) cluster |
| CMzTab::CMMzTabStream | |
| CCmpHypothesesByScore | |
| CCmpMassTraceByMZ | |
| CColorBrewer | |
| ►CTargetedSpectraExtractor::Comparator | |
| CTargetedSpectraExtractor::BinnedSpectrumComparator | |
| CMapAlignmentAlgorithmSpectrumAlignment::Compare | Inner class necessary for using the sort algorithm |
| CAccurateMassSearchEngine::CompareEntryAndMass_ | |
| CCompareTypeImpl< AAcid, char > | |
| CCompareTypeImpl< AAcid, uint8_t > | |
| CCompomer | Holds information on an edge connecting two features from a (putative) charge ladder |
| CMRMFeatureQC::ComponentGroupPairQCs | Quality Controls (QCs) for multiple components (between or within component_groups) |
| CMRMFeaturePicker::ComponentGroupParams | Structure to contain information about a component group with its parameters |
| CMRMFeatureQC::ComponentGroupQCs | Quality Controls (QCs) within a component group |
| CMRMFeaturePicker::ComponentParams | Structure to contain information about a single component with its parameters |
| CMRMFeatureQC::ComponentQCs | Quality Controls (QCs) for individual components |
| CSiriusMSFile::CompoundInfo | |
| CMetaboTargetedAssay::CompoundTargetDecoyPair | CompoundTargetDecoyPair stores a pair of CompoundInfo and MSSpectrum (target, decoy) |
| CConnectedComponent | |
| CConsensusMapNormalizerAlgorithmMedian | Algorithms of ConsensusMapNormalizer |
| CConsensusMapNormalizerAlgorithmQuantile | Algorithms of ConsensusMapNormalizer |
| CConsensusMapNormalizerAlgorithmThreshold | Algorithms of ConsensusMapNormalizer |
| CConsoleUtils | |
| CAASequence::ConstIterator | ConstIterator for AASequence |
| CNASequence::ConstIterator | ConstIterator of NASequence class |
| CConstRefVector< ContainerT > | This vector holds pointer to the elements of another container |
| ►CConstRefVector< ContainerT >::ConstRefVectorConstIterator< ValueT > | ConstIterator for the ConstRefVector |
| CConstRefVector< ContainerT >::ConstRefVectorIterator< ValueT > | Mutable iterator for the ConstRefVector |
| CMSExperiment::ContainerAdd_< ContainerValueType, addMassTraces > | Helper class to add either general data points in set2DData or use mass traces from meta values |
| CMSExperiment::ContainerAdd_< ContainerValueType, false > | |
| CMSExperiment::ContainerAdd_< ContainerValueType, true > | |
| CRawMSSignalSimulation::ContaminantInfo | |
| CContaminants::ContaminantsSummary | Structure for storing results |
| ►CContinuousWaveletTransform | This class is the base class of the continuous wavelet transformation |
| CContinuousWaveletTransformNumIntegration | This class computes the continuous wavelet transformation using a marr wavelet |
| CControlledVocabulary | Representation of a controlled vocabulary |
| CConvexHull2D | A 2-dimensional hull representation in [counter]clockwise direction - depending on axis labelling |
| COPXLDataStructs::CrossLinkSpectrumMatch | The CrossLinkSpectrumMatch struct represents a PSM between a ProteinProteinCrossLink and a spectrum in OpenPepXL |
| CCsiFingerIdMzTabWriter::CsiAdapterHit | Internal structure used in SiriusAdapter that is used for the conversion of the Csi:FingerID output to an mzTab |
| CCsiFingerIdMzTabWriter::CsiAdapterIdentification | |
| CCsiFingerIdMzTabWriter::CsiAdapterRun | |
| CCsiFingerIdMzTabWriter | |
| CCubicSpline2d | Cubic spline interpolation as described in R.L. Burden, J.D. Faires, Numerical Analysis, 4th ed. PWS-Kent, 1989, ISBN 0-53491-585-X, pp. 126-131 |
| CCV | |
| CCVMappingRule | Representation of a CV Mapping rule used by CVMappings |
| CCVMappings | Representation of controlled vocabulary mapping rules (for PSI formats) |
| CCVMappingTerm | Representation of controlled vocabulary term |
| CCVReference | Controlled Vocabulary Reference |
| CControlledVocabulary::CVTerm | Representation of a CV term |
| CCVTerm | Representation of controlled vocabulary term |
| CSemanticValidator::CVTerm | Representation of a parsed CV term |
| CEGHFitter1D::Data | Helper struct (contains the size of an area and a raw data container) |
| CEmgFitter1D::Data | Helper struct (contains the size of an area and a raw data container) |
| COptimizePeakDeconvolution::Data | Class containing the data needed for optimization |
| COptimizePick::Data | |
| CTwoDOptimization::Data | Helper struct (contains the size of an area and a raw data container) |
| CMzIdentMLDOMHandler::DatabaseInput | Struct to hold the information from the DatabaseInput xml tag |
| CDataFilters::DataFilter | Representation of a peak/feature filter combining FilterType, FilterOperation and a value (either double or String) |
| CDataFilters | DataFilter array providing some convenience functions |
| CTransformationModel::DataPoint | Coordinate pair (with optional annotation) |
| ►CDataTabBase | All tabs need to implement this interface |
| CDIATreeTab | Hierarchical visualization and selection of spectra |
| CSpectraIDViewTab | Tabular visualization / selection of identified spectra |
| CSpectraTreeTab | Hierarchical visualization and selection of spectra |
| CDataValue | Class to hold strings, numeric values, lists of strings and lists of numeric values |
| CDateTime | DateTime Class |
| CDaTrait | |
| CMzIdentMLDOMHandler::DBSequence | Struct to hold the information from the DBSequence xml tag |
| CDecoyGenerator | Methods to generate isobaric decoy sequences for DDA target-decoy searches |
| CDecoyHelper | Helper class for calculations on decoy proteins |
| ►Cdefault_dfs_visitor | |
| CIDBoostGraph::dfs_ccsplit_visitor | A boost dfs visitor that copies connected components into a vector of graphs |
| ►CDefaultHandler | |
| ►CXMLHandler | Base class for XML handlers |
| CCVMappingFile | Used to load CvMapping files |
| CConsensusXMLFile | This class provides Input functionality for ConsensusMaps and Output functionality for alignments and quantitation |
| CFeatureXMLFile | This class provides Input/Output functionality for feature maps |
| CIdXMLFile | Used to load and store idXML files |
| CMascotXMLHandler | Handler that is used for parsing MascotXML data |
| CMzDataHandler | |
| CMzIdentMLHandler | XML STREAM handler for MzIdentMLFile |
| ►CMzMLHandler | Handler for mzML file format |
| ►CMSDataWritingConsumer | Consumer class that writes MS data to disk using the mzML format |
| CNoopMSDataWritingConsumer | Consumer class that perform no operation |
| CPlainMSDataWritingConsumer | Consumer class that writes MS data to disk using the mzML format |
| CMzQuantMLHandler | XML handler for MzQuantMLFile |
| CMzXMLHandler | |
| CPTMXMLHandler | Handler that is used for parsing PTMXML data |
| ►CParamXMLHandler | XML Handler for Param files |
| CToolDescriptionHandler | XML handler for ToolDescriptionFile |
| ►CSemanticValidator | Semantically validates XML files using CVMappings and a ControlledVocabulary |
| CMzDataValidator | Semantically validates MzXML files |
| CMzIdentMLValidator | Semantically validates MzXML files |
| CMzMLValidator | Semantically validates MzXML files |
| CMzQuantMLValidator | Semantically validates MzQuantML files |
| CTraMLValidator | Semantically validates MzXML files |
| CTraMLHandler | XML handler for TraMLFile |
| CUnimodXMLHandler | Handler that is used for parsing XTandemXML data |
| CXQuestResultXMLHandler | XMLHandler for the result files of XQuest |
| COMSSAXMLFile | Used to load OMSSAXML files |
| CPepXMLFile | Used to load and store PepXML files |
| CPepXMLFileMascot | Used to load Mascot PepXML files |
| CProtXMLFile | Used to load (storing not supported, yet) ProtXML files |
| CQcMLFile | File adapter for QcML files used to load and store QcML files |
| CTransformationXMLFile | Used to load and store TransformationXML files |
| CXTandemXMLFile | Used to load XTandemXML files |
| ►CDefaultParamHandler | A base class for all classes handling default parameters |
| ►CBaseModel< 1 > | |
| ►CInterpolationModel | Abstract class for 1D-models that are approximated using linear interpolation |
| CBiGaussModel | BiGaussian distribution approximated using linear interpolation |
| CEGHModel | Exponential-Gaussian hybrid distribution model for elution profiles |
| CEmgModel | Exponentially modified gaussian distribution model for elution profiles |
| CExtendedIsotopeModel | Extended isotope distribution approximated using linear interpolation |
| CGaussModel | Normal distribution approximated using linear interpolation |
| CIsotopeModel | Isotope distribution approximated using linear interpolation |
| ►CBaseModel< 2 > | |
| CProductModel< 2 > | The class template is only implemented for D=2 because we use Peak2D here |
| ►CSignalToNoiseEstimator< MSSpectrum > | |
| CSignalToNoiseEstimatorMeanIterative< Container > | Estimates the signal/noise (S/N) ratio of each data point in a scan based on an iterative scheme which discards high intensities |
| CSignalToNoiseEstimatorMedian< Container > | Estimates the signal/noise (S/N) ratio of each data point in a scan by using the median (histogram based) |
| CAScore | Implementation of the Ascore For a given peptide sequence and its MS/MS spectrum it identifies the most probable phosphorylation-site(s). For each phosphorylation site a probability score is calculated. The algorithm is implemented according to Beausoleil et al. (Nat. Biotechnol. 2006) |
| CAbsoluteQuantitation | AbsoluteQuantitation is a class to support absolute or relative quantitation for targeted or untargeted quantitation workflows (e.g., Isotope Dilution Mass Spectrometry) |
| CAccurateMassSearchEngine | An algorithm to search for exact mass matches from a spectrum against a database (e.g. HMDB) |
| ►CBaseGroupFinder | The base class of all element group finding algorithms |
| CLabeledPairFinder | The LabeledPairFinder allows the matching of labeled features (features with a fixed distance) |
| CQTClusterFinder | A variant of QT clustering for the detection of feature groups |
| CSimplePairFinder | This class implements a simple point pair finding algorithm |
| CStablePairFinder | This class implements a pair finding algorithm for consensus features |
| ►CBaseLabeler | Abstract base class for all kinds of labeling techniques |
| CICPLLabeler | Simulate ICPL experiments |
| CITRAQLabeler | Simulate iTRAQ experiments |
| CLabelFreeLabeler | Abstract base class for all kinds of labeling techniques |
| CO18Labeler | Simulate O-18 experiments |
| CSILACLabeler | Simulate SILAC experiments |
| CBaseModel< D > | Abstract base class for all D-dimensional models |
| ►CBaseSuperimposer | The base class of all superimposer algorithms |
| CPoseClusteringAffineSuperimposer | A superimposer that uses a voting scheme, also known as pose clustering, to find a good affine transformation |
| CPoseClusteringShiftSuperimposer | A superimposer that uses a voting scheme, also known as pose clustering, to find a good shift transformation |
| CBasicProteinInferenceAlgorithm | Algorithm class that implements simple protein inference by aggregation of peptide scores. It has multiple parameter options like the aggregation method, when to distinguish peptidoforms, and if you want to use shared peptides ("use_shared_peptides"). First, the best PSM per spectrum is used, then only the best PSM per peptidoform is aggregated. Peptidoforms can optionally be distinguished via the treat_X_separate parameters: |
| CBayesianProteinInferenceAlgorithm | Performs a Bayesian protein inference on Protein/Peptide identifications or ConsensusMap (experimental) |
| CBernNorm | BernNorm scales the peaks by ranking them and then scaling them according to rank |
| ►CBinnedSpectrumCompareFunctor | Base class for compare functors of BinnedSpectra |
| CBinnedSharedPeakCount | Compare functor scoring the shared peaks for similarity measurement |
| CBinnedSpectralContrastAngle | Compare functor scoring the spectral contrast angle for similarity measurement |
| CBinnedSumAgreeingIntensities | Sum of agreeing intensities for similarity measurement |
| ►CCompNovoIdentificationBase | Run with CompNovoIdentificationBase |
| CCompNovoIdentification | Run with CompNovoIdentification |
| CCompNovoIdentificationCID | Run with CompNovoIdentificationCID |
| ►CCompNovoIonScoringBase | Run with CompNovoIonScoringBase |
| CCompNovoIonScoring | Run with CompNovoIonScoring |
| CCompNovoIonScoringCID | Run with CompNovoIonScoringCID |
| ►CConsensusIDAlgorithm | Abstract base class for all ConsensusID algorithms (that calculate a consensus from multiple ID runs) |
| ►CConsensusIDAlgorithmIdentity | Abstract base class for ConsensusID algorithms that compare only identical sequences |
| CConsensusIDAlgorithmAverage | Calculates a consensus from multiple ID runs by averaging the search scores |
| CConsensusIDAlgorithmBest | Calculates a consensus from multiple ID runs by taking the best search score |
| CConsensusIDAlgorithmRanks | Calculates a consensus from multiple ID runs based on the ranks of the search hits |
| CConsensusIDAlgorithmWorst | Calculates a consensus from multiple ID runs by taking the worst search score (conservative approach) |
| ►CConsensusIDAlgorithmSimilarity | Abstract base class for ConsensusID algorithms that take peptide similarity into account |
| CConsensusIDAlgorithmPEPIons | Calculates a consensus from multiple ID runs based on PEPs and shared ions |
| CConsensusIDAlgorithmPEPMatrix | Calculates a consensus from multiple ID runs based on PEPs and sequence similarities |
| CConsensusMapMergerAlgorithm | Merges identification data in ConsensusMaps |
| CDBSuitability | This class holds the functionality of calculating the database suitability |
| CDIAScoring | Scoring of an spectrum at the peak apex of an chromatographic elution peak |
| CDeNovoAlgorithm | Base class for ion scoring implementation for de novo algorithms |
| CDeNovoIdentification | Base class for de novo identification |
| CDeNovoIonScoring | Base class for ion scoring implementation for de novo algorithms |
| CDeNovoPostScoring | Base class for ion scoring implementation for de novo algorithms |
| CDetectabilitySimulation | Simulates peptide detectability |
| CDiaPrescore | Scoring of an spectrum given library intensities of a transition group |
| CDigestSimulation | Simulates protein digestion |
| CElutionModelFitter | Helper class for fitting elution models to features |
| CElutionPeakDetection | Extracts chromatographic peaks from a mass trace |
| CEmgGradientDescent | Compute the area, background and shape metrics of a peak |
| CFIAMSDataProcessor | Data processing for FIA-MS data |
| CFalseDiscoveryRate | Calculates false discovery rates (FDR) from identifications |
| CFeatureDeconvolution | An algorithm to decharge features (i.e. as found by FeatureFinder) |
| CFeatureDistance | A functor class for the calculation of distances between features or consensus features |
| ►CFeatureFinderAlgorithm | Abstract base class for FeatureFinder algorithms |
| CFeatureFinderAlgorithmIsotopeWavelet | Implements the isotope wavelet feature finder |
| CFeatureFinderAlgorithmMRM | FeatureFinderAlgorithm for MRM experiments |
| CFeatureFinderAlgorithmPicked | FeatureFinderAlgorithm for picked peaks |
| CFeatureFinderAlgorithmMetaboIdent | |
| CFeatureFinderIdentificationAlgorithm | |
| CFeatureFinderMultiplexAlgorithm | |
| CFeatureFindingMetabo | Method for the assembly of mass traces belonging to the same isotope pattern, i.e., that are compatible in retention times, mass-to-charge ratios, and isotope abundances |
| ►CFeatureGroupingAlgorithm | Base class for all feature grouping algorithms |
| CFeatureGroupingAlgorithmKD | A feature grouping algorithm for unlabeled data |
| CFeatureGroupingAlgorithmLabeled | A map feature grouping algorithm for labeling techniques with two labels |
| CFeatureGroupingAlgorithmQT | A feature grouping algorithm for unlabeled data |
| CFeatureGroupingAlgorithmUnlabeled | A map feature grouping algorithm for unlabeled data |
| ►CFilterFunctor | A FilterFunctor extracts some spectrum characteristics for quality assessment |
| CComplementFilter | Total intensity of peak pairs that could result from complementing fragments of charge state 1 |
| CGoodDiffFilter | GoodDiffFilter counts the number ob peak pairs whose m/z difference can be explained by a amino acid loss |
| CIntensityBalanceFilter | IntensityBalanceFilter divides the m/z-range into ten regions and sums the intensity in these regions |
| CIsotopeDiffFilter | IsotopeDiffFilter returns total intensity of peak pairs that could result from isotope peaks |
| CNeutralLossDiffFilter | NeutralLossDiffFilter returns the total intensity ob peak pairs whose m/z difference can be explained by a neutral loss |
| CTICFilter | TICFilter calculates TIC |
| ►CFitter1D | Abstract base class for all 1D-dimensional model fitter |
| ►CLevMarqFitter1D | Abstract class for 1D-model fitter using Levenberg-Marquardt algorithm for parameter optimization |
| CEGHFitter1D | Exponential-Gaussian hybrid distribution fitter (1-dim.) using Levenberg-Marquardt algorithm (Eigen implementation) for parameter optimization |
| CEmgFitter1D | Exponentially modified gaussian distribution fitter (1-dim.) using Levenberg-Marquardt algorithm (Eigen implementation) for parameter optimization |
| ►CMaxLikeliFitter1D | Abstract base class for all 1D-model fitters using maximum likelihood optimization |
| CBiGaussFitter1D | BiGaussian distribution fitter (1-dim.) approximated using linear interpolation |
| CExtendedIsotopeFitter1D | Extended isotope distribution fitter (1-dim.) approximated using linear interpolation |
| CGaussFitter1D | Gaussian distribution fitter (1-dim.) approximated using linear interpolation |
| CIsotopeFitter1D | Isotope distribution fitter (1-dim.) approximated using linear interpolation |
| CGaussFilter | This class represents a Gaussian lowpass-filter which works on uniform as well as on non-uniform profile data |
| CIDDecoyProbability | IDDecoyProbability calculates probabilities using decoy approach |
| CIDMapper | Annotates an MSExperiment, FeatureMap or ConsensusMap with peptide identifications |
| CIDMergerAlgorithm | Creates a new Protein ID run into which other runs can be inserted. Creates union of protein hits but concatenates PSMs. Checks search engine consistency of all inserted runs. It differs from the IDMerger tool, in that it is an algorithm class and it allows inserting multiple peptide hits per peptide sequence (not only the first occurrence) |
| CIDRipper | Ripping protein/peptide identification according their file origin |
| CIDScoreSwitcherAlgorithm | |
| CInclusionExclusionList | Provides functionality for writing inclusion or exclusion lists |
| CIonizationSimulation | Simulates Protein ionization |
| CIsobaricChannelExtractor | Extracts individual channels from MS/MS spectra for isobaric labeling experiments |
| CIsobaricQuantifier | Given the extracted channel intensities the IsobaricQuantifier corrects and normalizes the intensities for further processing |
| ►CIsobaricQuantitationMethod | Abstract base class describing an isobaric quantitation method in terms of the used channels and an isotope correction matrix |
| CItraqEightPlexQuantitationMethod | ITRAQ 8 plex quantitation to be used with the IsobaricQuantitation |
| CItraqFourPlexQuantitationMethod | ITRAQ 4 plex quantitation to be used with the IsobaricQuantitation |
| CTMTElevenPlexQuantitationMethod | TMT 11plex quantitation to be used with the IsobaricQuantitation |
| CTMTSixPlexQuantitationMethod | TMT 6plex quantitation to be used with the IsobaricQuantitation |
| CTMTSixteenPlexQuantitationMethod | TMT 16plex quantitation to be used with the IsobaricQuantitation |
| CTMTTenPlexQuantitationMethod | TMT 10plex quantitation to be used with the IsobaricQuantitation |
| CIsotopeLabelingMDVs | IsotopeLabelingMDVs is a class to support and analyze isotopic labeling experiments (i.e. MDVs : Mass Distribution Vectors, also known as Mass Isotopomer Distribution (MID)) |
| CKDTreeFeatureMaps | Stores a set of features, together with a 2D tree for fast search |
| ►CLinearResampler | Linear Resampling of raw data |
| CLinearResamplerAlign | Linear Resampling of raw data with alignment |
| CLowessSmoothing | LOWESS (locally weighted scatterplot smoothing) |
| CMRMDecoy | This class generates a TargetedExperiment object with decoys based on a TargetedExperiment object |
| CMRMFeatureFilter | The MRMFeatureFilter either flags components and/or transitions that do not pass the QC criteria or filters out components and/or transitions that do not pass the QC criteria |
| CMRMFeatureFinderScoring | The MRMFeatureFinder finds and scores peaks of transitions that co-elute |
| CMRMFragmentSelection | This class can select appropriate fragment ions of an MS/MS spectrum of a peptide |
| CMRMMapping | A class to map targeted assays to chromatograms |
| CMRMTransitionGroupPicker | The MRMTransitionGroupPicker finds peaks in chromatograms that belong to the same precursors |
| CMSPFile | File adapter for MSP files (NIST spectra library) |
| CMSPGenericFile | Load MSP text file and save it into an `MSExperiment` |
| CMSSim | Central class for simulation of mass spectrometry experiments |
| CMapAlignmentAlgorithmIdentification | A map alignment algorithm based on peptide identifications from MS2 spectra |
| CMapAlignmentAlgorithmPoseClustering | A map alignment algorithm based on pose clustering |
| CMapAlignmentAlgorithmSpectrumAlignment | A map alignment algorithm based on spectrum similarity (dynamic programming) |
| CMapAlignmentAlgorithmTreeGuided | A map alignment algorithm based on peptide identifications from MS2 spectra |
| CMarkerMower | MarkerMower uses PeakMarker to find peaks, those that are not marked get removed |
| CMascotGenericFile | Read/write Mascot generic files (MGF) |
| CMascotRemoteQuery | Class which handles the communication between OpenMS and the Mascot server |
| CMassDecompositionAlgorithm | Mass decomposition algorithm, given a mass it suggests possible compositions |
| CMassTraceDetection | A mass trace extraction method that gathers peaks similar in m/z and moving along retention time |
| CMasstraceCorrelator | Correlates individual masstraces found in mass spectrometric maps |
| CPosteriorErrorProbabilityModel | Implements a mixture model of the inverse gumbel and the gauss distribution or a gaussian mixture |
| CMetaboliteFeatureDeconvolution | An algorithm to decharge small molecule features (i.e. as found by FeatureFinder) |
| CMetaboliteSpectralMatching | |
| CMorphologicalFilter | This class implements baseline filtering operations using methods from mathematical morphology |
| CMultiplexDeltaMassesGenerator | Generates complete list of all possible mass shifts due to isotopic labelling |
| CNLargest | NLargest removes all but the n largest peaks |
| CNormalizer | Normalizes the peak intensities spectrum-wise |
| CNucleicAcidSpectrumGenerator | Generates theoretical spectra for nucleic acid sequences |
| COfflinePrecursorIonSelection | Implements different algorithms for precursor ion selection |
| COpenPepXLAlgorithm | Search for peptide pairs linked with a labeled cross-linker |
| COpenPepXLLFAlgorithm | Search for cross-linked peptide pairs in tandem MS spectra |
| COptimizePeakDeconvolution | This class provides the deconvolution of peak regions using non-linear optimization |
| CPSLPFormulation | Implements ILP formulation of precursor selection problems |
| CParentPeakMower | ParentPeakMower gets rid of high peaks that could stem from unfragmented precursor ions |
| CPeakIntegrator | Compute the area, background and shape metrics of a peak |
| ►CPeakMarker | PeakMarker marks peaks that seem to fulfill some criterion |
| CComplementMarker | ComplementMarker marks peak pairs which could represent y - b ion pairs |
| CIsotopeMarker | IsotopeMarker marks peak pairs which could represent an ion and its isotope |
| CNeutralLossMarker | NeutralLossMarker marks peak pairs which could represent an ion an its neutral loss (water, ammonia) |
| CPeakPickerCWT | This class implements a peak picking algorithm using wavelet techniques |
| CPeakPickerHiRes | This class implements a fast peak-picking algorithm best suited for high resolution MS data (FT-ICR-MS, Orbitrap). In high resolution data, the signals of ions with similar mass-to-charge ratios (m/z) exhibit little or no overlapping and therefore allow for a clear separation. Furthermore, ion signals tend to show well-defined peak shapes with narrow peak width |
| CPeakPickerIterative | This class implements a peak-picking algorithm for high-resolution MS data (specifically designed for TOF-MS data) |
| CPeakPickerMRM | The PeakPickerMRM finds peaks a single chromatogram |
| CPeakPickerSH | |
| ►CPeakSpectrumCompareFunctor | Base class for compare functors of spectra, that return a similarity value for two spectra |
| CPeakAlignment | Make a PeakAlignment of two PeakSpectra |
| CSpectraSTSimilarityScore | Similarity score of SpectraST |
| CSpectrumAlignmentScore | Similarity score via spectra alignment |
| CSpectrumCheapDPCorr | SpectrumCheapDPCorr calculates an optimal alignment on stick spectra |
| CSpectrumPrecursorComparator | SpectrumPrecursorComparator compares just the parent mass of two spectra |
| CSteinScottImproveScore | Similarity score based of Stein & Scott |
| CZhangSimilarityScore | Similarity score of Zhang |
| CPeptideAndProteinQuant | Helper class for peptide and protein quantification based on feature data annotated with IDs |
| CPeptideIndexing | Refreshes the protein references for all peptide hits in a vector of PeptideIdentifications and adds target/decoy information |
| ►CPlotCanvas | Base class for visualization canvas classes |
| CPlot1DCanvas | Canvas for visualization of one or several spectra |
| CPlot2DCanvas | Canvas for 2D-visualization of peak map, feature map and consensus map data |
| CPlot3DCanvas | Canvas for 3D-visualization of peak map data |
| CPrecursorIonSelection | This class implements different precursor ion selection strategies |
| CPrecursorIonSelectionPreprocessing | This class implements the database preprocessing needing for precursor ion selection |
| CProteinResolver | Helper class for peptide and protein quantification based on feature data annotated with IDs |
| CProtonDistributionModel | A proton distribution model to calculate the proton distribution over charged peptides |
| CQuantitativeExperimentalDesign | Merge files according to experimental design |
| CRTSimulation | Simulates/Predicts retention times for peptides or peptide separation |
| CRawMSSignalSimulation | Simulates MS signals for a given set of peptides |
| CRawTandemMSSignalSimulation | Simulates tandem MS signals for a given set of peptides |
| CSONARScoring | Scoring of an spectrum using SONAR data |
| CSavitzkyGolayFilter | Computes the Savitzky-Golay filter coefficients using QR decomposition |
| CScaler | Scaler scales the peak by ranking the peaks and assigning intensity according to rank |
| ►CSignalToNoiseEstimator< Container > | This class represents the abstract base class of a signal to noise estimator |
| CSignalToNoiseEstimatorMedian< OpenMS::MSChromatogram > | |
| CSignalToNoiseEstimatorMedian< ContainerT > | |
| CSimpleSVM | Simple interface to support vector machines for classification (via LIBSVM) |
| CSimpleSearchEngineAlgorithm | |
| CSimpleTSGXLMS | Generates theoretical spectra for cross-linked peptides |
| CSiriusAdapterAlgorithm | |
| CSpectraIDViewTab | Tabular visualization / selection of identified spectra |
| CSpectraMerger | Merges blocks of MS or MS2 spectra |
| CSpectraMerger::SpectraDistance_ | |
| CSpectrumAlignment | Aligns the peaks of two sorted spectra Method 1: Using a banded (width via 'tolerance' parameter) alignment if absolute tolerances are given. Scoring function is the m/z distance between peaks. Intensity does not play a role! |
| CSpectrumAnnotator | Annotates spectra from identifications and theoretical spectra or identifications from spectra and theoretical spectra matching with various options |
| CSqrtMower | Scales the intensity of peaks to the sqrt |
| CSvmTheoreticalSpectrumGenerator | Simulates MS2 spectra with support vector machines |
| CSvmTheoreticalSpectrumGeneratorTrainer | Train SVM models that are used by SvmTheoreticalSpectrumGenerator |
| CSwathMapMassCorrection | A class containing correction functions for Swath MS maps |
| CSwathWizardBase | Main window of the SwathWizard tool |
| CTOFCalibration | This class implements an external calibration for TOF data using external calibrant spectra |
| CTOPPASBase | Main window of the TOPPAS tool |
| CTOPPViewBase | Main window of TOPPView tool |
| CTargetedSpectraExtractor | This class filters, annotates, picks, and scores spectra (e.g., taken from a DDA experiment) based on a target list |
| CTheoreticalSpectrumGenerator | Generates theoretical spectra for peptides with various options |
| CTheoreticalSpectrumGeneratorXLMS | Generates theoretical spectra for cross-linked peptides |
| CThresholdMower | ThresholdMower removes all peaks below a threshold |
| ►CTraceFitter | Abstract fitter for RT profile fitting |
| CEGHTraceFitter | A RT Profile fitter using an Exponential Gaussian Hybrid background model |
| CGaussTraceFitter | Fitter for RT profiles using a Gaussian background model |
| ►CTransitionTSVFile | This class supports reading and writing of OpenSWATH transition lists |
| CTransitionPQPFile | This class supports reading and writing of PQP files |
| CTwoDOptimization | This class provides the two-dimensional optimization of the picked peak parameters |
| CWindowMower | WindowMower augments the highest peaks in a sliding or jumping window |
| CXFDRAlgorithm | Calculates false discovery rate estimates on crosslink identifications |
| CDeisotoper | |
| CMultiplexDeltaMasses::DeltaMass | Mass shift with corresponding label set |
| CSvmTheoreticalSpectrumGenerator::DescriptorSet | A set of descriptors for a single training row |
| ►CDigestionEnzyme | Base class for digestion enzymes |
| CDigestionEnzymeProtein | Representation of a digestion enzyme for proteins (protease) |
| CDigestionEnzymeRNA | Representation of a digestion enzyme for RNA (RNase) |
| CDigestionEnzymeDB< DigestionEnzymeType, InstanceType > | Digestion enzyme database (base class) |
| ►CDigestionEnzymeDB< DigestionEnzymeProtein, ProteaseDB > | |
| CProteaseDB | Database for enzymes that digest proteins (proteases) |
| ►CDigestionEnzymeDB< DigestionEnzymeRNA, RNaseDB > | |
| CRNaseDB | Database for enzymes that digest RNA (RNases) |
| CIDFilter::DigestionFilter | Is peptide evidence digestion product of some protein |
| ►CDIntervalBase< D > | A base class for D-dimensional interval |
| CDBoundingBox< 1 > | |
| CDBoundingBox< 2 > | |
| CDRange< 1 > | |
| CDRange< 2 > | |
| CDRange< 3 > | |
| CDBoundingBox< D > | A D-dimensional bounding box |
| CDRange< D > | A D-dimensional half-open interval |
| CDistanceMatrix< Value > | A two-dimensional distance matrix, similar to OpenMS::Matrix |
| CFeatureDistance::DistanceParams_ | Structure for storing distance parameters |
| CColorBrewer::Distinct | |
| ►CDocumentIdentifier | Manage source document information |
| CConsensusMap | A container for consensus elements |
| ►CExperimentalSettings | Description of the experimental settings |
| CMSExperiment | In-Memory representation of a mass spectrometry experiment |
| CMSQuantifications | |
| CFeatureMap | A container for features |
| CDocumentIDTagger | Tags OpenMS file containers with a DocumentID |
| CListUtils::DoubleTolerancePredicate_ | Predicate to check double equality with a given tolerance |
| CDPeak< dimensions > | Metafunction to choose among Peak1D respectively Peak2D through a template argument |
| CDPosition< D, TCoordinateType > | Representation of a coordinate in D-dimensional space |
| CDPosition< 1 > | |
| CDPosition< 2 > | |
| CDPosition< 2, Int64 > | |
| CDPosition< DIMENSION > | |
| CDTAFile | File adapter for DTA files |
| CEDTAFile | File adapter for Enhanced DTA files |
| CElement | Representation of an element |
| CQTCluster::Element | |
| CElementDB | Singleton that stores elements |
| CEmgGradientDescent_friend | |
| CEmgScoring | Scoring of an elution peak using an exponentially modified gaussian distribution model |
| CEmpiricalFormula | Representation of an empirical formula |
| ►CEnhancedTabBarWidgetInterface | Widgets that are placed into an EnhancedTabBar must implement this interface |
| ►CPlotWidget | Base class for spectrum widgets |
| CPlot1DWidget | Widget for visualization of several spectra |
| CPlot2DWidget | Widget for 2D-visualization of peak map and feature map data |
| CPlot3DWidget | Widget for 3D-visualization of map data |
| CTOPPASWidget | Widget visualizing and allowing to edit TOPP pipelines |
| ►CEnzymaticDigestion | Class for the enzymatic digestion of sequences |
| CProteaseDigestion | Class for the enzymatic digestion of proteins |
| CRNaseDigestion | Class for the enzymatic digestion of RNAs |
| CEnzymaticDigestionLogModel | Class for the Log L model of enzymatic digestion of proteins |
| CEqualInTolerance< CompareType > | Struct for binary predicate to consider equality with a certain tolerance |
| ►CErrorHandler | |
| CXMLValidator | Validator for XML files |
| CEuclideanSimilarity | CompareFunctor for 2Dpoints |
| ►Cexception | STL class |
| ►Cbad_alloc | STL class |
| COutOfMemory | Out of memory exception |
| ►Cruntime_error | STL class |
| ►CBaseException | Exception base class |
| CClusterFunctor::InsufficientInput | Exception thrown if not enough data (<2) is used |
| CBufferOverflow | Buffer overflow exception |
| CConversionError | Invalid conversion exception |
| CDepletedIDPool | Exception used if no more unique document ID's can be drawn from ID pool |
| CDivisionByZero | Division by zero error exception |
| CElementNotFound | Element could not be found exception |
| CFailedAPICall | A call to an external library (other than OpenMS) went wrong |
| CFileEmpty | File is empty |
| CFileNameTooLong | Filename is too long to be writable/readable by the filesystem |
| CFileNotFound | File not found exception |
| CFileNotReadable | File not readable exception |
| CFileNotWritable | File not writable exception |
| CIOException | General IOException |
| CIllegalArgument | A method or algorithm argument contains illegal values |
| CIllegalPosition | Invalid 3-dimensional position exception |
| CIllegalSelfOperation | Illegal self operation exception |
| CIllegalTreeOperation | Illegal tree operation exception |
| CIncompatibleIterators | Incompatible iterator exception |
| CIndexOverflow | Int overflow exception |
| CIndexUnderflow | Int underflow exception |
| CInvalidIterator | Invalid iterator exception |
| CInvalidParameter | Exception indicating that an invalid parameter was handed over to an algorithm |
| CInvalidRange | Invalid range exception |
| CInvalidSize | Invalid UInt exception |
| CInvalidValue | Invalid value exception |
| CMissingInformation | Not all required information provided |
| CNotImplemented | Not implemented exception |
| CNullPointer | Null pointer argument is invalid exception |
| COutOfGrid | Out of grid exception |
| COutOfMemory | Out of memory exception |
| COutOfRange | Out of range exception |
| CParseError | Parse Error exception |
| CPostcondition | Postcondition failed exception |
| CPrecondition | Precondition failed exception |
| CRequiredParameterNotGiven | A required parameter was not given |
| CSizeUnderflow | UInt underflow exception |
| CSqlOperationFailed | SqlOperation failed exception |
| CUnableToCalibrate | Exception used if an error occurred while calibrating a dataset |
| CUnableToCreateFile | Unable to create file exception |
| CUnableToFit | Exception used if an error occurred while fitting a model to a given dataset |
| CUnregisteredParameter | An unregistered parameter was accessed |
| CWrongParameterType | A parameter was accessed with the wrong type |
| CFeatureFinderDefs::NoSuccessor | Exception that is thrown if a method an invalid IndexPair is given |
| CXMLHandler::EndParsingSoftly | Exception that is thrown if the parsing is ended by some event (e.g. if only a prefix of the XML file is needed) |
| CMap< Key, T >::IllegalKey | Map illegal key exception |
| CUnnormalizedComparator | Exception thrown if clustering is attempted without a normalized compare functor |
| CExperimentalDesign | Representation of an experimental design in OpenMS. Instances can be loaded with the ExperimentalDesignFile class |
| CExperimentalDesignFile | Load an experimental design from a TSV file. (see ExperimentalDesign for details on the supported format) |
| CExternalProcessMBox | A wrapper around ExternalProcess to conveniently show a MessageBox when an error occurs |
| CChromatogramExtractorAlgorithm::ExtractionCoordinates | |
| ►CFactoryBase | Base class for Factory<T> |
| CFactory< FactoryProduct > | Returns FactoryProduct* based on the name of the desired concrete FactoryProduct |
| CFAIMSHelper | Helper functions for FAIMS data |
| CFASTAContainer< TBackend > | Template parameter for vector-based FASTA access |
| CFASTAContainer< TFI_File > | FASTAContainer<TFI_File> will make FASTA entries available chunk-wise from start to end by loading it from a FASTA file. This avoids having to load the full file into memory. While loading, the container will memorize the file offsets of each entry, allowing to read an arbitrary i'th entry again from disk. If possible, only entries from the currently cached chunk should be queried, otherwise access will be slow |
| CFASTAContainer< TFI_Vector > | FASTAContainer<TFI_Vector> simply takes an existing vector of FASTAEntries and provides the same interface with a potentially huge speed benefit over FASTAContainer<TFI_File> since it does not need disk access, but at the cost of memory |
| CFASTAFile::FASTAEntry | FASTA entry type (identifier, description and sequence) The first String corresponds to the identifier that is written after the > in the FASTA file. The part after the first whitespace is stored in description and the text from the next line until the next > (exclusive) is stored in sequence |
| CFeatureFinderAlgorithmMetaboIdent::FeatureCompare | Comparison functor for features |
| CFeatureFinderIdentificationAlgorithm::FeatureCompare | Comparison functor for features |
| CAbsoluteQuantitationStandards::featureConcentration | Structure to hold a single component and its corresponding known concentration |
| CFeatureFileOptions | Options for loading files containing features |
| CFeatureFinderIdentificationAlgorithm::FeatureFilterPeptides | Predicate for filtering features by assigned peptides: |
| CFeatureFinderAlgorithmMetaboIdent::FeatureFilterQuality | Predicate for filtering features by overall quality |
| CFeatureFinderIdentificationAlgorithm::FeatureFilterQuality | Predicate for filtering features by overall quality: |
| CFeatureFinderAlgorithmPickedHelperStructs | Wrapper struct for all the classes needed by the FeatureFinderAlgorithmPicked and the associated classes |
| ►CFeatureFinderDefs | The purpose of this struct is to provide definitions of classes and typedefs which are used throughout all FeatureFinder classes |
| CFeatureFinder | The main feature finder class |
| CFeatureFinderAlgorithmMRM | FeatureFinderAlgorithm for MRM experiments |
| CFeatureFinderAlgorithmPicked | FeatureFinderAlgorithm for picked peaks |
| CFitter1D | Abstract base class for all 1D-dimensional model fitter |
| CFeatureFinderAlgorithmMetaboIdent::FeatureFinderMetaboIdentCompound | Compound in the assay library |
| CFeatureHypothesis | Internal structure used in FeatureFindingMetabo that keeps track of a feature hypothesis (isotope group hypothesis) |
| CFeatureMapping | |
| CFeatureMapping::FeatureMappingInfo | Stores information required for preprocessing |
| CFeatureMapping::FeatureToMs2Indices | Stores preprocessed feature mapping information |
| CIsotopeWavelet::fi_ | Internal union for fast computation of the power function |
| CFIAMSScheduler | |
| CFile | Basic file handling operations |
| CFileHandler | Facilitates file handling by file type recognition |
| CFileMapping | Maps input/output files to filenames for the external program |
| CFileTypes::FileTypeList | Holds a vector of known file types, e.g. as a way to specify supported input formats |
| CFileTypes | Centralizes the file types recognized by FileHandler |
| CFlagSet< ENUM > | Stores and handles combinations of enum values, e.g. a set of flags as bits flipped in an UInt64 |
| CFlagSet< LayerData::DataType > | |
| CFlagSet< TV_STATUS > | |
| CPeptideIndexing::FoundProteinFunctor | |
| CRNPxlFragmentAnnotationHelper::FragmentAnnotationDetail_ | Single fragment annotation |
| CGumbelMaxLikelihoodFitter::Functor< _Scalar, NX, NY > | |
| ►CGumbelMaxLikelihoodFitter::Functor< double > | |
| CGumbelMaxLikelihoodFitter::GumbelDistributionFunctor | |
| CFuzzyStringComparator | Fuzzy comparison of strings, tolerates numeric differences |
| CGammaDistributionFitter::GammaDistributionFitResult | Struct to represent the parameters of a gamma distribution |
| CGammaDistributionFitter | Implements a fitter for the Gamma distribution |
| CGaussFilterAlgorithm | This class represents a Gaussian lowpass-filter which works on uniform as well as on non-uniform profile data |
| CGaussFitter::GaussFitResult | Struct of parameters of a Gaussian distribution |
| CGaussFitter | Implements a fitter for Gaussian functions |
| CSignalToNoiseEstimator< Container >::GaussianEstimate | Protected struct to store parameters my, sigma for a Gaussian distribution |
| ►CLevMarqFitter1D::GenericFunctor | |
| CEGHFitter1D::EGHFitterFunctor | |
| CEmgFitter1D::EgmFitterFunctor | |
| ►CTraceFitter::GenericFunctor | |
| CEGHTraceFitter::EGHTraceFunctor | |
| CGaussTraceFitter::GaussTraceFunctor | |
| CIDFilter::GetMatchingItems< HitType, Entry > | Builds a map index of data that have a String index to find matches and return the objects |
| CIDFilter::GetMatchingItems< OpenMS::PeptideEvidence, OpenMS::FASTAFile::FASTAEntry > | |
| CConfidenceScoring::GLM_ | Binomial GLM |
| CGlobalExceptionHandler | OpenMS global exception handler |
| CGradient | Representation of a HPLC gradient |
| CGridBasedCluster | Basic data structure for clustering |
| CGridFeature | Representation of a feature in a hash grid |
| CGridSearch< TupleTypes > | |
| CGUILock | RAII class to disable the GUI and set a busy cursor and go back to the orignal state when this class is destroyed |
| CGumbelDistributionFitter::GumbelDistributionFitResult | Struct to represent the parameters of a gumbel distribution |
| CGumbelMaxLikelihoodFitter::GumbelDistributionFitResult | Struct to represent the parameters of a gumbel distribution |
| CGumbelDistributionFitter | Implements a fitter for the Gumbel distribution |
| CGumbelMaxLikelihoodFitter | Implements a fitter for the Gumbel distribution |
| CGzipIfstream | Decompresses files which are compressed in the gzip format (*.gzip) |
| CHasActivationMethod< SpectrumType > | Predicate that determines if a spectrum was generated using any activation method given in the constructor list |
| CIDFilter::HasDecoyAnnotation< HitType > | Is this a decoy hit? |
| CIDFilter::HasGoodScore< HitType > | Is the score of this hit at least as good as the given value? |
| Chash< OpenMS::String > | |
| CHashGrid< Cluster > | Container for (2-dimensional coordinate, value) pairs |
| CNucleicAcidSearchEngine::HasInvalidLength | |
| CIDFilter::HasMatchingAccession< HitType > | Given a list of protein accessions, do any occur in the annotation(s) of this hit? |
| CIDFilter::HasMatchingAccessionUnordered< HitType > | Given a list of protein accessions, do any occur in the annotation(s) of this hit? |
| CIDFilter::HasMaxMetaValue< HitType > | Does a meta value of this hit have at most the given value? |
| CIDFilter::HasMaxRank< HitType > | Is the rank of this hit below or at the given cut-off? |
| CHasMetaValue< MetaContainer > | Predicate that determines if a class has a certain metavalue |
| CIDFilter::HasMetaValue< HitType > | Is a meta value with given key and value set on this hit? |
| CIDFilter::HasNoHits< IdentificationType > | Is the list of hits of this peptide/protein ID empty? |
| CHasPrecursorCharge< SpectrumType > | Predicate that determines if a spectrum has a certain precursor charge as given in the constructor list |
| CHasScanMode< SpectrumType > | Predicate that determines if a spectrum has a certain scan mode |
| CHasScanPolarity< SpectrumType > | Predicate that determines if a spectrum has a certain scan polarity |
| CHDF5Connector | File adapter for HDF5 files |
| CHiddenMarkovModel | Hidden Markov Model implementation of PILIS |
| CHistogram< ValueType, BinSizeType > | Representation of a histogram |
| CConsensusIDAlgorithm::HitInfo | |
| CHMMState | Hidden Markov Model State class for the Hidden Markov Model |
| CHost< Pattern< TNeedle, FuzzyAC > > | |
| CHost< Pattern< TNeedle, FuzzyAC > const > | |
| CHPLC | Representation of a HPLC experiment |
| CHyperScore | An implementation of the X!Tandem HyperScore PSM scoring function |
| CIBSpectraFile | Implements the export of consensusmaps into the IBSpectra format used by isobar to load quantification results |
| CIChromatogramsReader | The interface of read-access to a list of chromatograms |
| CIChromatogramsWriter | |
| ►CIDataFrameWriter | |
| CCSVWriter | |
| CDataMatrix | |
| CIDBoostGraph | Creates and maintains a boost graph based on the OpenMS ID datastructures |
| CIDConflictResolverAlgorithm | Resolves ambiguous annotations of features with peptide identifications |
| CIdentificationDataConverter | |
| CMs2IdentificationRate::IdentificationRateData | Structure for storing results |
| CIDFilter | Collection of functions for filtering peptide and protein identifications |
| CMzTab::IDMzTabStream | |
| CIDScoreGetterSetter | A class for extracting and reinserting IDScores from Peptide/ProteinIdentifications and from ConsensusMaps |
| CInclusionExclusionList::IEWindow | |
| ►CIFeature | |
| CFeatureOpenMS | An implementation of the OpenSWATH Feature Access interface using OpenMS |
| CMockFeature | Mock object implementing IFeature |
| CILPDCWrapper | |
| CIMDataConverter | This class converts PeakMaps and MSSpectra from/to different IM/FAIMS storage models |
| ►CIMRMFeature | |
| CMRMFeatureOpenMS | An implementation of the OpenSWATH MRM Feature Access interface using OpenMS |
| CMockMRMFeature | Mock object implementing IMRMFeature |
| CIMSAlphabet | Holds an indexed list of bio-chemical elements |
| ►CIMSAlphabetParser< AlphabetElementType, Container, InputSource > | An abstract templatized parser to load the data that is used to initialize Alphabet objects |
| CIMSAlphabetTextParser | Implements abstract AlphabetParser to read data from the plain text format |
| ►CIMSDataConsumer | The interface of a consumer of spectra and chromatograms |
| ►CFullSwathFileConsumer | Abstract base class which can consume spectra coming from SWATH experiment stored in a single file |
| CCachedSwathFileConsumer | On-disk cached implementation of FullSwathFileConsumer |
| CMzMLSwathFileConsumer | On-disk mzML implementation of FullSwathFileConsumer |
| CRegularSwathFileConsumer | In-memory implementation of FullSwathFileConsumer |
| CMSDataAggregatingConsumer | Aggregates spectra by retention time |
| CMSDataCachedConsumer | Transforming and cached writing consumer of MS data |
| CMSDataChainingConsumer | Consumer class that passes all consumed data through a set of operations |
| CMSDataSqlConsumer | A data consumer that inserts MS data into a SQLite database |
| CMSDataStoringConsumer | Consumer class that simply stores the data |
| CMSDataTransformingConsumer | Transforming consumer of MS data |
| CMSDataWritingConsumer | Consumer class that writes MS data to disk using the mzML format |
| CNoopMSDataConsumer | Consumer class that performs no operation |
| CIMSElement | Represents a chemical atom with name and isotope distribution |
| CIMSIsotopeDistribution | Represents a distribution of isotopes restricted to the first K elements |
| CIMTypes | |
| CIndexedMzMLDecoder | A class to analyze indexedmzML files and extract the offsets of individual tags |
| CIndexedMzMLFileLoader | A class to load an indexedmzML file |
| CIndexedMzMLHandler | A low-level class to read an indexedmzML file |
| CFeatureHandle::IndexLess | Comparator by map and unique id |
| CPSLPFormulation::IndexLess | |
| CPSLPFormulation::IndexTriple | Struct that holds the indices of the precursors in the feature map and the ilp formulation |
| CInIntensityRange< PeakType > | Predicate that determines if a peak lies inside/outside a specific intensity range |
| CINIUpdater | |
| CInMSLevelRange< SpectrumType > | Predicate that determines if a spectrum lies inside/outside a specific MS level set |
| CInMzRange< PeakType > | Predicate that determines if a peak lies inside/outside a specific m/z range |
| CInPrecursorMZRange< SpectrumType > | Predicate that determines if a spectrum's precursor is within a certain m/z range |
| CFuzzyStringComparator::InputLine | Stores information about the current input line (i.e., stream for the line and the current position in the stream) |
| ►CInputSource | |
| CCompressedInputSource | This class is based on xercesc::LocalFileInputSource |
| CInRTRange< SpectrumType > | Predicate that determines if a spectrum lies inside/outside a specific retention time range |
| CInspectInfile | Inspect input file adapter |
| CInspectOutfile | Representation of an Inspect outfile |
| CChromatogramPeak::IntensityLess | Comparator by intensity |
| CPeak1D::IntensityLess | |
| CPeak2D::IntensityLess | |
| CTransformationModelInterpolated::Interpolator | The class defines a generic interpolation technique used in the TransformationModelInterpolated |
| CTOPPASToolVertex::IOInfo | Stores the information for input/output files/lists |
| CIonMobilityScoring | A class that calls the ion mobility scoring routines |
| CCompNovoIonScoringBase::IonScore | |
| CDeNovoIonScoring::IonScore | IonScore |
| CSvmTheoreticalSpectrumGenerator::IonType | Nested class |
| ►Cios_base | STL class |
| ►Cbasic_ios< Char > | STL class |
| ►Cbasic_istream< Char > | STL class |
| ►Cbasic_ifstream< Char > | STL class |
| ►Cifstream | STL class |
| CFidHandler | Read-only fid File handler for XMass Analysis |
| ►Cbasic_ostream< Char > | STL class |
| ►Costream | STL class |
| CLogStream | Log Stream Class |
| CSVOutStream | Stream class for writing to comma/tab/...-separated values files |
| CProteinResolver::ISDGroup | |
| CIsEmptySpectrum< SpectrumType > | Predicate that determines if a spectrum is empty |
| CIDScoreGetterSetter::IsHitType< T > | |
| CIDScoreGetterSetter::IsIDType< T > | |
| ►CISignalToNoise | |
| CSignalToNoiseOpenMS< ContainerT > | An implementation of the OpenSWATH SignalToNoise Access interface using OpenMS |
| CMockSignalToNoise | Mock object implementing ISignalToNoise |
| CIsInCollisionEnergyRange< SpectrumType > | Predicate that determines if an MSn spectrum was generated with a collision energy in the given range |
| CIsInIsolationWindow< SpectrumType > | Predicate that determines if the isolation window covers ANY of the given m/z values |
| CIsInIsolationWindowSizeRange< SpectrumType > | Predicate that determines if the width of the isolation window of an MSn spectrum is in the given range |
| CIsobaricQuantitationMethod::IsobaricChannelInformation | Summary of an isobaric quantitation channel |
| CIsobaricIsotopeCorrector | Performs isotope impurity correction on the intensities extracted from an isobaric labeling experiment |
| CIsobaricNormalizer | Performs median normalization on the extracted ratios of isobaric labeling experiment |
| CIsobaricQuantifierStatistics | Statistics for quantitation performance and comparison of NNLS vs. naive method (aka matrix inversion) |
| ►CIsoSpecGeneratorWrapper | Interface for the IsoSpec algorithm - a generator of infinitely-resolved theoretical spectra |
| CIsoSpecOrderedGeneratorWrapper | Generate the stream of configurations, ordered from most likely to least likely |
| CIsoSpecThresholdGeneratorWrapper | Provides a threshold-based generator of isotopologues: generates all isotopologues more probable than a given probability threshold |
| CIsoSpecTotalProbGeneratorWrapper | Generate a p-set of configurations for a given p (that is, a set of configurations such that their probabilities sum up to p). The p in normal usage will usually be close to 1 (e.g. 0.99) |
| ►CIsoSpecWrapper | A convenience class for the IsoSpec algorithm - easier to use than the IsoSpecGeneratorWrapper classes |
| CIsoSpecThresholdWrapper | A non-generator version of IsoSpecThresholdGeneratorWrapper |
| CIsoSpecTotalProbWrapper | Create a p-set of configurations for a given p (that is, a set of configurations such that their probabilities sum up to p). The p in normal usage will usually be close to 1 (e.g. 0.99) |
| CIsotopeCluster | Stores information about an isotopic cluster (i.e. potential peptide charge variants) |
| CIsotopeDistribution | |
| CIsotopeDistributionCache | Pre-calculate isotope distributions for interesting mass ranges |
| CFeatureFinderAlgorithmPickedHelperStructs::IsotopePattern | Helper structure for a found isotope pattern used in FeatureFinderAlgorithmPicked |
| ►CIsotopePatternGenerator | Provides an interface for different isotope pattern generator methods |
| CCoarseIsotopePatternGenerator | Isotope pattern generator for coarse isotope distributions |
| CFineIsotopePatternGenerator | Isotope pattern generator for fine isotope distributions |
| CIsotopeWavelet | Implements the isotope wavelet function |
| CIsotopeWaveletTransform< PeakType > | A class implementing the isotope wavelet transform. If you just want to find features using the isotope wavelet, take a look at the FeatureFinderAlgorithmIsotopeWavelet class. Usually, you only have to consider the class at hand if you plan to change the basic implementation of the transform |
| CISpectraReader | The interface of read-access to a list of spectra |
| CISpectraWriter | |
| ►CISpectrumAccess | The interface of a mass spectrometry experiment |
| CSpectrumAccessOpenMS | An implementation of the OpenSWATH Spectrum Access interface using OpenMS |
| CSpectrumAccessOpenMSCached | An implementation of the Spectrum Access interface using on-disk caching |
| CSpectrumAccessOpenMSInMemory | An implementation of the OpenSWATH Spectrum Access interface completely in memory |
| CSpectrumAccessSqMass | An implementation of the Spectrum Access interface using SQL files |
| ►CSpectrumAccessTransforming | An abstract base class implementing a transforming wrapper around spectrum access |
| CSpectrumAccessQuadMZTransforming | A transforming m/z wrapper around spectrum access using a quadratic equation |
| CIsZoomSpectrum< SpectrumType > | Predicate that determines if a spectrum is a zoom (enhanced resolution) spectrum |
| ►CIterator | |
| CIteratorWrapper< Iterator > | Wrapper that adds operator< to iterators, so they can be used as (part of) keys in maps/sets or multi_index_containers |
| CAASequence::Iterator | Iterator class for AASequence |
| CNASequence::Iterator | Iterator of NASequence class |
| ►Citerator | |
| CHashGrid< Cluster >::ConstIterator | Constant element iterator for the hash grid |
| CHashGrid< Cluster >::Iterator | Element iterator for the hash grid |
| CAreaIterator< ValueT, ReferenceT, PointerT, SpectrumIteratorT, PeakIteratorT > | Forward iterator for an area of peaks in an experiment |
| CIntensityIteratorWrapper< IteratorT > | An iterator wrapper to access peak intensities instead of the peak itself |
| ►Cset< K >::iterator | STL iterator class |
| CIteratorWrapper< DataQueries::iterator > | |
| CIteratorWrapper< DataProcessingSoftwares::iterator > | |
| ►CITransitionGroup | |
| CTransitionGroupOpenMS< SpectrumT, TransitionT > | An implementation of the OpenSWATH Transition Group Access interface using OpenMS |
| CMockTransitionGroup | Mock object implementing ITransitionGroup |
| CItraqConstants | Some constants used throughout iTRAQ classes |
| CJavaInfo | Detect Java and retrieve information |
| CKDTreeFeatureNode | A node of the kD-tree with pointer to corresponding data and index |
| CKroenikFile | File adapter for Kroenik (HardKloer sibling) files |
| CMultiplexDeltaMassesGenerator::Label | Complete label information |
| ►CLayerAnnotatorBase | |
| CLayerAnnotatorAMS | |
| CLayerAnnotatorOSW | |
| CLayerAnnotatorPeptideID | |
| CLayerData | Class that stores the data for one layer |
| CLayerStack | |
| CLexicographicComparator< Cmp1, Cmp2 > | A wrapper class that combines two comparators lexicographically. Normally you should use the make-function lexicographicComparator() because then you do not need to specify the template arguments |
| CLibSVMEncoder | Serves for encoding sequences into feature vectors |
| CLightCompound | |
| CLightModification | |
| CLightProtein | |
| CLightTargetedExperiment | |
| CLightTransition | |
| CLinearInterpolation< Key, Value > | Provides access to linearly interpolated values (and derivatives) from discrete data points. Values beyond the given range of data points are implicitly taken as zero |
| CLinearInterpolation< double > | |
| CLinearRegression | This class offers functions to perform least-squares fits to a straight line model, 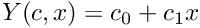 |
| CLinearRegressionWithoutIntercept | This class offers functions to perform least-squares fits to a straight line model, |
| ►Clist< T > | STL class |
| CAnnotations1DContainer | Container for annotations to content of Plot1DCanvas |
| CListUtils | Collection of utility functions for management of vectors |
| CLocalLinearMap::LLMParam | Define parameters needed by the Local Linear Map (LLM) model |
| CLocalLinearMap | Trained Local Linear Map (LLM) model for peak intensity prediction |
| CInternalCalibration::LockMass | Helper class, describing a lock mass |
| CLogStreamBuf::LogCacheStruct | Holds a counter of occurrences and an index for the occurrence sequence of the corresponding log message |
| CLogConfigHandler | The LogConfigHandler provides the functionality to configure the internal logging of OpenMS algorithms that use the global instances of LogStream |
| CLogStreamNotifier | |
| CSimpleTSGXLMS::LossIndex | |
| CTheoreticalSpectrumGeneratorXLMS::LossIndex | |
| CLPWrapper | |
| CPercolatorFeatureSetHelper::lq_PeptideEvidence | For accession dependent sorting of PeptideEvidences |
| CPercolatorFeatureSetHelper::lq_ProteinHit | For accession dependent sorting of ProteinHits |
| ►Cmap< K, T > | STL class |
| CMap< QString, QList< OpenMS::TOPPASResource > > | |
| CMap< UInt, std::vector< const OpenMS::ResidueModification * > > | |
| CMap< OpenMS::String, UInt > | |
| CMap< OpenMS::String, OpenMS::Map< OpenMS::String, UInt > > | |
| CMap< OpenMS::String, OpenMS::SourceFile > | |
| CMap< OpenMS::String, OpenMS::String > | |
| CMap< OpenMS::String, OpenMS::Software > | |
| CMap< Size, OpenMS::String > | |
| CMap< OpenMS::String, IntList > | |
| CMap< Size, std::set< OpenMS::String > > | |
| CMap< const OpenMS::Residue *, char > | |
| CMap< OpenMS::String, std::vector< DataProcessingPtr > > | |
| CMap< OpenMS::String, std::pair< OpenMS::String, OpenMS::String > > | |
| CMap< OpenMS::HMMState *, OpenMS::Map< OpenMS::HMMState *, std::vector< double > > > | |
| CMap< PointType::CoordinateType, DBoundingBox< 1 > > | |
| CMap< OpenMS::String, std::vector< OpenMS::CVMappingRule > > | |
| CMap< OpenMS::String, OpenMS::AASequence > | |
| CMap< OpenMS::HMMState *, OpenMS::Map< OpenMS::HMMState *, double > > | |
| CMap< OpenMS::String, const DigestionEnzymeType * > | |
| CMap< Size, std::vector< double > > | |
| CMap< std::pair< OpenMS::String, OpenMS::String >, bool > | |
| CMap< OpenMS::String, Size > | |
| CMap< OpenMS::String, std::set< OpenMS::String > > | |
| CMap< Size, OpenMS::Map< Size, std::set< OpenMS::String > > > | |
| CMap< OpenMS::HMMState *, OpenMS::Map< OpenMS::HMMState *, Size > > | |
| CMap< OpenMS::HMMState *, OpenMS::Map< OpenMS::HMMState *, std::pair< OpenMS::HMMState *, OpenMS::HMMState * > > > | |
| CMap< OpenMS::HMMState *, std::vector< double > > | |
| CMap< UInt, UInt > | |
| CMap< OpenMS::HMMState *, std::set< OpenMS::HMMState * > > | |
| CMap< OpenMS::EnzymaticDigestionLogModel::BindingSite_, OpenMS::EnzymaticDigestionLogModel::CleavageModel_ > | |
| CMap< OpenMS::String, OpenMS::ControlledVocabulary::CVTerm > | |
| CMap< OpenMS::String, std::vector< OpenMS::CVTerm > > | |
| CMap< OpenMS::String, OpenMS::Map< OpenMS::String, OpenMS::Map< OpenMS::String, UInt > > > | |
| CMap< Size, Size > | |
| CMap< OpenMS::String, OpenMS::Sample > | |
| CMap< OpenMS::String, std::vector< OpenMS::Internal::SemanticValidator::CVTerm > > | |
| CMap< OpenMS::String, OpenMS::HMMState * > | |
| CMap< QString, QString > | |
| CMap< double, std::vector< OpenMS::MassDecomposition > > | |
| CMap< OpenMS::String, OpenMS::Instrument > | |
| CMap< OpenMS::HMMState *, std::pair< OpenMS::HMMState *, OpenMS::HMMState * > > | |
| CMap< OpenMS::String, OpenMS::Map< OpenMS::String, std::pair< OpenMS::String, OpenMS::String > > > | |
| CMap< OpenMS::HMMState *, double > | |
| CMap< char, const OpenMS::Residue * > | |
| CMap< OpenMS::HMMState *, Size > | |
| CMap< char, double > | |
| CMap< Key, T > | Map class based on the STL map (containing several convenience functions) |
| CMapAlignmentAlgorithmKD | An efficient reference-free feature map alignment algorithm for unlabeled data |
| ►CMapAlignmentEvaluationAlgorithm | Base class for all Caap evaluation algorithms |
| CMapAlignmentEvaluationAlgorithmPrecision | Caap evaluation algorithm to obtain a precision value |
| CMapAlignmentEvaluationAlgorithmRecall | Caap evaluation algorithm to obtain a recall value |
| CMapAlignmentTransformer | This class collects functions for applying retention time transformations to data structures |
| CMapConversion | |
| CProteinIdentification::Mapping | Two way mapping from ms-run-path to protID|pepID-identifier |
| CAccurateMassSearchEngine::MappingEntry_ | |
| CMappingParam | Filename mappings for all input/output files |
| CConsensusFeature::MapsLess | Compare by the sets of consensus elements (lexicographically) |
| CModifiedPeptideGenerator::MapToResidueType | |
| CMapUtilities< MapType > | Utilities for Feature and ConsensusMap |
| ►CMapUtilities< ConsensusMap > | |
| CConsensusMap | A container for consensus elements |
| ►CMapUtilities< FeatureMap > | |
| CFeatureMap | A container for features |
| CMassDecomposer< ValueType, DecompositionValueType > | An interface to handle decomposing of integer values/masses over a set of integer weights (alphabet) |
| ►CMassDecomposer< long unsigned int, unsigned int > | |
| CIntegerMassDecomposer< ValueType, DecompositionValueType > | Implements MassDecomposer interface using algorithm and data structures described in paper "Efficient Mass Decomposition" S. Böcker, Z. Lipták, ACM SAC-BIO, 2004 doi:10.1145/1066677.1066715 |
| CMassDecomposition | Class represents a decomposition of a mass into amino acids |
| CMassExplainer | Computes empirical formulas for given mass differences using a set of allowed elements |
| CIMSAlphabet::MassSortingCriteria_ | Private class-functor to sort out elements in mass ascending order |
| CFeatureFinderAlgorithmPickedHelperStructs::MassTrace | Helper struct for mass traces used in FeatureFinderAlgorithmPicked |
| CMassTrace | A container type that gathers peaks similar in m/z and moving along retention time |
| CFeatureFinderAlgorithmMetaboIdent::MassTraceBounds | Boundaries for a mass trace in a feature |
| CTargetedSpectraExtractor::Match | |
| CMatchedIterator< CONT_T, TRAIT, CONST_T > | For each element in the reference container the closest peak in the target will be searched. If no match is found within the tolerance window, the peak will be skipped over |
| Cmean_and_stddev | Functor to compute the mean and stddev of sequence using the std::foreach algorithm |
| CSysInfo::MemUsage | A convenience class to report either absolute or delta (between two timepoints) RAM usage |
| CMessagePasserFactory< Label > | |
| CMetaboTargetedTargetDecoy::MetaboTargetDecoyMassMapping | MetaboTargetDecoyMassMapping introduces a mapping of target and decoy masses and their respective compound reference using an identifier |
| CMetaboTargetedAssay | This class provides methods for the extraction of targeted assays for metabolomics |
| CMetaboTargetedTargetDecoy | Resolve overlapping fragments and missing decoys for experimental specific decoy generation in targeted/pseudo targeted metabolomics |
| CMetaInfo | A Type-Name-Value tuple class |
| ►CMetaInfoInterface | Interface for classes that can store arbitrary meta information (Type-Name-Value tuples) |
| CAcquisition | Information about one raw data spectrum that was combined with several other raw data spectra |
| CAcquisitionInfo | Description of the combination of raw data to a single spectrum |
| ►CCVTermList | Representation of controlled vocabulary term list |
| CIncludeExcludeTarget | This class stores a SRM/MRM transition |
| CPrecursor | Precursor meta information |
| CProduct | Product meta information |
| CReactionMonitoringTransition | This class stores a SRM/MRM transition |
| ►CSoftware | Description of the software used for processing |
| CDataProcessingSoftware | Information about software used for data processing |
| CSourceFile | Description of a file location, used to store the origin of (meta) data |
| CConfiguration | |
| CContact | |
| CInstrument | |
| ►CPeptideCompound | Base class to represent either a peptide or a compound |
| CCompound | Represents a compound (small molecule) |
| CPeptide | Represents a peptide (amino acid sequence) |
| CPrediction | |
| CProtein | |
| CPublication | |
| ►CCVTermListInterface | Interface to the controlled vocabulary term list |
| CInterpretation | Product ion interpretation |
| CPeptide::Modification | |
| CRetentionTime | This class stores a retention time structure that is used in TargetedExperiment (representing a TraML file) |
| CTraMLProduct | Represents a product ion |
| ►CChromatogramSettings | Representation of chromatogram settings, e.g. SRM/MRM chromatograms |
| CMSChromatogram | The representation of a chromatogram |
| CConsensusMap | A container for consensus elements |
| CConsensusMap::ColumnHeader | Description of the columns in a consensus map |
| CContactPerson | Contact person information |
| CDataProcessing | Description of the applied preprocessing steps |
| CExperimentalSettings | Description of the experimental settings |
| CFeatureMap | A container for features |
| CIdentification | Represents a object which can store the information of an analysisXML instance |
| CIdentificationData | Representation of spectrum identification results and associated data |
| CDBSearchParam | Parameters specific to a database search step |
| CDataProcessingStep | Data processing step that is applied to the data (e.g. database search, PEP calculation, filtering, ConsensusID) |
| CDataQuery | Search query, e.g. spectrum or feature |
| CMoleculeParentMatch | Meta data for the association between an identified molecule (e.g. peptide) and a parent molecule (e.g. protein) |
| CScoreType | Information about a score type |
| ►CScoredProcessingResult | Base class for ID data with scores and processing steps (and meta info) |
| CIdentifiedCompound | |
| CIdentifiedSequence< SeqType > | Identified sequence (peptide or oligonucleotide) |
| CMoleculeQueryMatch | Meta data for a search hit (e.g. peptide-spectrum match) |
| CParentMolecule | Representation of a parent molecule that is identified only indirectly (e.g. a protein) |
| CParentMoleculeGrouping | Set of groups of ambiguously identified parent molecules (e.g. results of running a protein inference algorithm) |
| CQueryMatchGroup | : Group of related (co-identified) molecule-query matches |
| CIdentificationHit | Represents a object which can store the information of an analysisXML instance |
| CInstrument | Description of a MS instrument |
| CInstrumentSettings | Description of the settings a MS Instrument was run with |
| CIonDetector | Description of a ion detector (part of a MS Instrument) |
| CIonSource | Description of an ion source (part of a MS Instrument) |
| CMassAnalyzer | Description of a mass analyzer (part of a MS Instrument) |
| ►CMetaInfoDescription | Description of the meta data arrays of MSSpectrum |
| CFloatDataArray | Float data array class |
| CIntegerDataArray | Integer data array class |
| CStringDataArray | String data array class |
| CPeptideHit | Representation of a peptide hit |
| CPeptideIdentification | Represents the peptide hits for a spectrum |
| CProteinHit | Representation of a protein hit |
| CProteinIdentification | Representation of a protein identification run |
| CProteinIdentification::SearchParameters | Search parameters of the DB search |
| ►CRichPeak2D | A 2-dimensional raw data point or peak with meta information |
| ►CBaseFeature | A basic LC-MS feature |
| CConsensusFeature | A consensus feature spanning multiple LC-MS/MS experiments |
| ►CFeature | An LC-MS feature |
| CMRMFeature | A multi-chromatogram MRM feature |
| CSample | Meta information about the sample |
| ►CSampleTreatment | Base class for sample treatments (Digestion, Modification, Tagging, ...) |
| CDigestion | Meta information about digestion of a sample |
| ►CModification | Meta information about chemical modification of a sample |
| CTagging | Meta information about tagging of a sample e.g. ICAT labeling |
| CScanWindow | Scan window description |
| CSpectrumIdentification | Represents a object which can store the information of an analysisXML instance |
| ►CSpectrumSettings | Representation of 1D spectrum settings |
| CMSSpectrum | The representation of a 1D spectrum |
| CMetaInfoInterfaceUtils | Utilities operating on containers inheriting from MetaInfoInterface |
| CMetaInfoRegistry | Registry which assigns unique integer indices to strings |
| CMetaKeyGetter< T > | |
| CLayerStatisticsDialog::MetaStatsValue_ | Struct representing the statistics about one meta information |
| CMinimumDistance | Basic data structure for distances between clusters |
| CTraceFitter::ModelData | |
| CModelDescription< D > | Stores the name and parameters of a model |
| CModificationDefinition | Representation of modification definition |
| CModificationDefinitionsSet | Representation of a set of modification definitions |
| CMzIdentMLDOMHandler::ModificationParam | Struct to hold the information from the ModificationParam xml tag |
| ►CModificationsDB | Database which holds all residue modifications from UniMod |
| CCrossLinksDB | |
| CModifiedNASequenceGenerator | |
| CModifiedPeptideGenerator | |
| CIdentificationData::ModifyMultiIndexAddProcessingStep< ElementType > | Helper functor for adding processing steps to elements in a boost::multi_index_container structure |
| CIdentificationData::ModifyMultiIndexAddScore< ElementType > | Helper functor for adding scores to elements in a boost::multi_index_container structure |
| CIdentificationData::ModifyMultiIndexRemoveParentMatches< ElementType > | Helper functor for removing invalid parent matches from elements in a boost::multi_index_container structure |
| CMorpheusScore | An implementation of the Morpheus PSM scoring function Inspired by a C# implementation by C. Wenger released under MIT license |
| CMQEvidence | Builds a MaxQuant Evidence.txt |
| CMRMBatchFeatureSelector | |
| CMRMFeaturePicker | _MRMFeaturePicker_ defines the structures containing parameters to be used in [MRMTransitionGroupPicker](MRMTransitionGroupPicker) for components and components groups |
| CMRMFeatureQC | The MRMFeatureQC is a class to handle the parameters and options for MRMFeatureFilter |
| ►CMRMFeatureSelector | |
| ►CMRMFeatureSelectorQMIP | |
| CMRMFeatureSelector_test | |
| CMRMFeatureSelectorScore | |
| CMRMIonSeries | Generate theoretical fragment ion series for use in MRMAssay and MRMDecoy |
| CMRMRTNormalizer | The MRMRTNormalizer will find retention time peptides in data |
| CMRMScoring | This class implements different scores for peaks found in SRM/MRM |
| CMRMTransitionGroup< ChromatogramType, TransitionType > | The representation of a group of transitions in a targeted proteomics experiment |
| CMRMTransitionGroup< SpectrumT, TransitionT > | |
| CProteinResolver::MSDGroup | Representation of an msd group. Contains peptides, proteins and a pointer to its ISD group |
| CExperimentalDesign::MSFileSectionEntry | |
| CMsInspectFile | File adapter for MsInspect files |
| CMSNumpressCoder | Class to encode and decode data encoded with MSNumpress |
| CMSPGenericFile_friend | |
| CMSstatsFile | File adapter for MSstats files |
| CMSstatsFile::MSstatsLine_ | |
| CMSstatsFile::MSstatsTMTLine_ | |
| CMultiGradient | A gradient of multiple colors and arbitrary distances between colors |
| CMultiplexDeltaMasses | Data structure for mass shift pattern |
| CMultiplexClustering::MultiplexDistance | Scaled Euclidean distance for clustering |
| CMultiplexFilteredMSExperiment | Data structure storing all peaks (and optionally their raw data points) of an experiment corresponding to one specific peak pattern |
| CMultiplexFilteredPeak | Data structure storing a single peak that passed all filters |
| CMultiplexIsotopicPeakPattern | Data structure for pattern of isotopic peaks |
| CMultiplexSatelliteCentroided | Data structure storing a single satellite peak |
| CMultiplexSatelliteProfile | Data structure storing a single satellite data point |
| CMzIdentMLDOMHandler | XML DOM handler for MzIdentMLFile |
| CMSChromatogram::MZLess | Comparator for the retention time |
| CPeak1D::MZLess | Comparator by m/z position |
| CPeak2D::MZLess | Comparator by m/z position |
| CMzMLHandlerHelper | Helper for mzML file format |
| CMzMLSpectrumDecoder | A class to decode input strings that contain an mzML chromatogram or spectrum tag |
| CMzMLSqliteHandler | Sqlite handler for storing spectra and chromatograms in sqMass format |
| CMzMLSqliteSwathHandler | Sqlite handler for SWATH data sets |
| CMzQCFile | File adapter for mzQC files used to load and store mzQC files |
| CMzTab | Data model of MzTab files. Please see the official MzTab specification at https://code.google.com/p/mztab/ |
| CMzTabAssayMetaData | |
| CMzTabBoolean | |
| CMzTabContactMetaData | |
| CMzTabCVMetaData | |
| CMzTabDouble | |
| CMzTabDoubleList | |
| CMzTabFile | File adapter for MzTab files |
| CMzTabInstrumentMetaData | |
| CMzTabInteger | |
| CMzTabIntegerList | |
| CMzTabMetaData | All meta data of a mzTab file. Please refer to specification for documentation |
| CMzTabModification | |
| CMzTabModificationList | |
| CMzTabModificationMetaData | |
| CMzTabMSRunMetaData | |
| CMzTabNucleicAcidSectionRow | NUC - Nucleic acid section (table-based) |
| CMzTabOligonucleotideSectionRow | OLI - Oligonucleotide section (table-based) |
| CMzTabOSMSectionRow | OSM - OSM (oligonucleotide-spectrum match) section (table-based) |
| CMzTabParameter | |
| CMzTabParameterList | |
| CMzTabPeptideSectionRow | PEP - Peptide section (Table based) |
| CMzTabProteinSectionRow | PRT - Protein section (Table based) |
| CMzTabPSMSectionRow | PSM - PSM section (Table based) |
| CMzTabSampleMetaData | |
| CMzTabSmallMoleculeSectionRow | SML Small molecule section (table based) |
| CMzTabSoftwareMetaData | |
| CMzTabSpectraRef | |
| CMzTabString | |
| CMzTabStringList | |
| CMzTabStudyVariableMetaData | |
| CMZTrafoModel | Create and apply models of a mass recalibration function |
| CReactionMonitoringTransition::NameLess | Comparator by name |
| CNASequence | Representation of a nucleic acid sequence |
| CSplineInterpolatedPeaks::Navigator | Iterator class for access of spline packages |
| CNeedlemanWunsch | This class serves for the calculation of the global alignment score of two amino acid sequences by using the Needleman-Wunsch-Algorithm. For match and mismatch it uses a similarity scoring matrix |
| CQTCluster::Neighbor | |
| CSignalToNoiseEstimatorMedianRapid::NoiseEstimator | Class to compute the noise value at a given position |
| C_Alloc_base< _Tp, _Alloc >::NoLeakAlloc | |
| CNonNegativeLeastSquaresSolver | Wrapper for a non-negative least squares (NNLS) solver |
| CMSNumpressCoder::NumpressConfig | Configuration class for MSNumpress |
| COMS_TranslateTableAAToChar_< T > | |
| COMS_TranslateTableByteToAA_< T > | |
| COMS_TranslateTableCharToAA_< T > | |
| COMSSACSVFile | File adapter for OMSSACSV files |
| COnDiscMSExperiment | Representation of a mass spectrometry experiment on disk |
| COpenMSBuildInfo | Struct with some static methods to get informations on the build configuration |
| COpenMSOSInfo | |
| COpenSwath_Ind_Scores | |
| COpenSwath_Scores | A structure to hold the different scores computed by OpenSWATH |
| COpenSwath_Scores_Usage | A structure to store which scores should be used by the OpenSWATH Algorithm |
| COpenSwathDataAccessHelper | Several helpers to convert OpenMS datastructures to structures that implement the OpenSWATH interfaces |
| COpenSwathHelper | A helper class that is used by several OpenSWATH tools |
| COpenSwathOSWWriter | Class to write out an OpenSwath OSW SQLite output (PyProphet input) |
| COpenSwathScoring | A class that calls the scoring routines |
| COpenSwathTSVWriter | Class to write out an OpenSwath TSV output (mProphet input) |
| COptimizePick | This class provides the non-linear optimization of the peak parameters |
| COptimizePick::OptPeakFunctor | |
| COPXLDataStructs | |
| COPXLHelper | Functions needed by OpenPepXL and OpenPepXLLF to reduce duplicated code |
| COPXLSpectrumProcessingAlgorithms | |
| COSBinaryDataArray | The datastructures used by the OpenSwath interfaces |
| COSChromatogram | A single chromatogram |
| COSChromatogramMeta | Identifying information for a chromatogram |
| COSSpectrum | The structure that captures the generation of a peak list (including the underlying acquisitions) |
| COSSpectrumMeta | Identifying information for a spectrum |
| COSWData | Holds all or partial information from an OSW file |
| COSWFile | This class serves for reading in and writing OpenSWATH OSW files |
| COSWHierarchy | Hierarchy levels of the OSWData tree |
| COSWIndexTrace | |
| COSWPeakGroup | |
| COSWPeptidePrecursor | A peptide with a charge state |
| COSWProtein | A Protein is the highest entity and contains one or more peptides which were found/traced |
| COSWTransition | High-level meta data of a transition |
| COverlapDetector | Given a set of levels (rows), try to add items at to topmost row which does not overlap an already placed item in this row (according to its x-coordinate) |
| CPairComparatorFirstElement< PairType > | Class for comparison of std::pair using first ONLY e.g. for use with std::sort |
| CPairComparatorFirstElementMore< PairType > | Class for comparison of std::pair using first ONLY e.g. for use with std::sort |
| CPairComparatorSecondElement< PairType > | Class for comparison of std::pair using second ONLY e.g. for use with std::sort |
| CPairComparatorSecondElementMore< PairType > | Class for comparison of std::pair using second ONLY e.g. for use with std::sort |
| CPairMatcherFirstElement< PairType > | Class for comparison of std::pair using first ONLY e.g. for use with std::sort |
| CPairMatcherSecondElement< PairType > | Struct for comparison of std::pair using second ONLY e.g. for use with std::sort |
| CParam | Management and storage of parameters / INI files |
| CParam::ParamEntry | Parameter entry used to store the actual information inside of a Param entry |
| CParameterInformation | Struct that captures all information of a command line parameter |
| CSiriusAdapterAlgorithm::ParameterModifier | |
| ►CSiriusAdapterAlgorithm::ParameterSection | |
| CSiriusAdapterAlgorithm::Fingerid | |
| CSiriusAdapterAlgorithm::Passatutto | |
| CSiriusAdapterAlgorithm::Preprocessing | |
| CSiriusAdapterAlgorithm::Project | |
| CSiriusAdapterAlgorithm::Sirius | |
| CParam::ParamIterator | Forward const iterator for the Param class |
| CParam::ParamNode | Node inside a Param object which is used to build the internal tree |
| CParamValue | Class to hold strings, numeric values, vectors of strings and vectors of numeric values using the stl types |
| CParentMoleculeGroup | : Group of ambiguously identified parent molecules (e.g. protein group) |
| CPattern< TNeedle, FuzzyAC > | |
| CPatternAuxData< TNeedle > | |
| CPatternAuxData< PeptideDB > | |
| CIMSIsotopeDistribution::Peak | Structure that represents an isotope peak - pair of mass and abundance |
| ►CPeak1D | A 1-dimensional raw data point or peak |
| CPrecursor | Precursor meta information |
| ►CPeak2D | A 2-dimensional raw data point or peak |
| ►CFeatureHandle | Representation of a Peak2D, RichPeak2D or Feature |
| CFeatureHandle::FeatureHandleMutable_ | Helper class returned by FeatureHandle::asMutable(), which see |
| CRichPeak2D | A 2-dimensional raw data point or peak with meta information |
| CPeptideHit::PeakAnnotation | Contains annotations of a peak |
| CPeakIntegrator::PeakArea | |
| CPeakPickerCWT::PeakArea_ | Class for the internal peak representation |
| CPeakIntegrator::PeakBackground | |
| CPeakPickerHiRes::PeakBoundary | Structure for peak boundaries |
| CPeakCandidate | A small structure to hold peak candidates |
| CPeakPickerMaxima::PeakCandidate | The PeakCandidate describes the output of the peak picker |
| CPeakFileOptions | Options for loading files containing peak data |
| CPeakIndex | Index of a peak or feature |
| CPeakIntensityPredictor | Predict peak heights of peptides based on Local Linear Map model |
| CPeakPickerMaxima | This class implements a fast peak-picking algorithm best suited for high resolution MS data (FT-ICR-MS, Orbitrap). In high resolution data, the signals of ions with similar mass-to-charge ratios (m/z) exhibit little or no overlapping and therefore allow for a clear separation. Furthermore, ion signals tend to show well-defined peak shapes with narrow peak width |
| CPeakShape | Internal representation of a peak shape (used by the PeakPickerCWT) |
| CPeakIntegrator::PeakShapeMetrics | |
| CPeakTypeEstimator | Estimates if the data of a spectrum is raw data or peak data |
| CPeakWidthEstimator | Rough estimation of the peak width at m/z |
| ►CPenaltyFactors | Class for the penalty factors used during the optimization |
| CPenaltyFactorsIntensity | Class for the penalty factors used during the optimization |
| CPepNovoInfile | PepNovo input file adapter |
| CPepNovoOutfile | Representation of a PepNovo output file |
| CPeptide | |
| CFeatureFinderIdentificationAlgorithm::PeptideCompare | Comparison functor for (unassigned) peptide IDs |
| CPeptideAndProteinQuant::PeptideData | Quantitative and associated data for a peptide |
| CIDFilter::PeptideDigestionFilter | Filter Peptide Hit by its digestion product |
| CProteinResolver::PeptideEntry | Peptide. First in silico. If experimental is set to true it is MS/MS derived |
| CMzIdentMLDOMHandler::PeptideEvidence | Struct to hold the PeptideEvidence information |
| CPeptideEvidence | Representation of a peptide evidence |
| COPXLHelper::PeptideIDScoreComparator | A comparator for PeptideIdentifications that compares the scores in the first PeptideHit |
| CPeptideIndexing::PeptideProteinMatchInformation | |
| CPeptideProteinResolution | Resolves shared peptides based on protein scores |
| CPeptideHit::PepXMLAnalysisResult | Analysis Result (containing search engine / prophet results) |
| COSWFile::PercolatorFeature | |
| CPercolatorFeatureSetHelper | Percolator feature set and integration helper |
| CPercolatorOutfile | Class for reading Percolator tab-delimited output files |
| CCompNovoIdentificationBase::Permut | Simple class to store permutations and a score |
| CPointerComparator< Cmp > | Wrapper that takes a comparator for `something' and makes a comparator for pointers to `something' out of it. Normally you should use the make-function pointerComparator() because then you do not need to specify the template arguments |
| CChromatogramPeak::PositionLess | Comparator by position. As this class has dimension 1, this is basically an alias for RTLess |
| CPeak1D::PositionLess | Comparator by position. As this class has dimension 1, this is basically an alias for MZLess |
| CPeak2D::PositionLess | Comparator by position. Lexicographical comparison (first RT then m/z) is done |
| CPeakShape::PositionLess | Comparison of mz_positions |
| CPpmTrait | |
| CPrecisionWrapper< FloatingPointType > | Wrapper class to implement output with appropriate precision. See precisionWrapper() |
| CPrecursorCorrection | This class provides methods for precursor correction |
| CNucleicAcidSearchEngine::PrecursorInfo | |
| CPrecursorMassComparator | |
| CPrecursorPurity | Precursor purity or noise estimation |
| CSimpleSVM::Prediction | SVM prediction result |
| CFuzzyStringComparator::PrefixInfo_ | Wrapper for the prefix information computed for the failure report |
| COPXLDataStructs::PreprocessedPairSpectra | The PreprocessedPairSpectra struct represents the result of comparing a light and a heavy labeled spectra to each other |
| CProbablePhosphoSites | |
| CProductModel< D > | Class for product models i.e. models with D independent dimensions |
| CReactionMonitoringTransition::ProductMZLess | Comparator by Product ion MZ |
| ►CProgressLogger | Base class for all classes that want to report their progress |
| CSignalToNoiseEstimator< MSSpectrum > | |
| CAccurateMassSearchEngine | An algorithm to search for exact mass matches from a spectrum against a database (e.g. HMDB) |
| CAverageLinkage | AverageLinkage ClusterMethod |
| CBaseGroupFinder | The base class of all element group finding algorithms |
| CBaseSuperimposer | The base class of all superimposer algorithms |
| CBasicProteinInferenceAlgorithm | Algorithm class that implements simple protein inference by aggregation of peptide scores. It has multiple parameter options like the aggregation method, when to distinguish peptidoforms, and if you want to use shared peptides ("use_shared_peptides"). First, the best PSM per spectrum is used, then only the best PSM per peptidoform is aggregated. Peptidoforms can optionally be distinguished via the treat_X_separate parameters: |
| CBayesianProteinInferenceAlgorithm | Performs a Bayesian protein inference on Protein/Peptide identifications or ConsensusMap (experimental) |
| CChromatogramExtractor | The ChromatogramExtractor extracts chromatograms from a spectra file |
| CChromatogramExtractorAlgorithm | The ChromatogramExtractorAlgorithm extracts chromatograms from a MS data |
| CCompleteLinkage | CompleteLinkage ClusterMethod |
| CConfidenceScoring | |
| CConsensusMapMergerAlgorithm | Merges identification data in ConsensusMaps |
| CConsensusXMLFile | This class provides Input functionality for ConsensusMaps and Output functionality for alignments and quantitation |
| CDTA2DFile | DTA2D File adapter |
| CElutionPeakDetection | Extracts chromatographic peaks from a mass trace |
| CFASTAFile | This class serves for reading in and writing FASTA files If the protein/gene sequence contains unusual symbols (such as translation end (*)), they will be kept! You can use aggregate methods load() and store() to read/write a set of protein sequences at the cost of memory |
| CFeatureFinder | The main feature finder class |
| CFeatureFinderMultiplexAlgorithm | |
| CFeatureFindingMetabo | Method for the assembly of mass traces belonging to the same isotope pattern, i.e., that are compatible in retention times, mass-to-charge ratios, and isotope abundances |
| CFeatureGroupingAlgorithmKD | A feature grouping algorithm for unlabeled data |
| CFeatureXMLFile | This class provides Input/Output functionality for feature maps |
| CGaussFilter | This class represents a Gaussian lowpass-filter which works on uniform as well as on non-uniform profile data |
| CGridBasedClustering< Metric > | 2D hierarchical clustering implementation optimized for large data sets containing many small clusters i.e. dimensions of clusters << dimension of entire dataset |
| CIDMergerAlgorithm | Creates a new Protein ID run into which other runs can be inserted. Creates union of protein hits but concatenates PSMs. Checks search engine consistency of all inserted runs. It differs from the IDMerger tool, in that it is an algorithm class and it allows inserting multiple peptide hits per peptide sequence (not only the first occurrence) |
| CIdXMLFile | Used to load and store idXML files |
| ►CCachedMzMLHandler | An class that uses on-disk caching to read and write spectra and chromatograms |
| CMSDataCachedConsumer | Transforming and cached writing consumer of MS data |
| CInternalCalibration | A mass recalibration method using linear/quadratic interpolation (robust/weighted) of given reference masses |
| CIonizationSimulation | Simulates Protein ionization |
| CLinearResampler | Linear Resampling of raw data |
| CMRMAssay | Generate assays from a TargetedExperiment |
| CMRMDecoy | This class generates a TargetedExperiment object with decoys based on a TargetedExperiment object |
| CMRMFeatureFinderScoring | The MRMFeatureFinder finds and scores peaks of transitions that co-elute |
| CMRMFeatureQCFile | File adapter for MRMFeatureQC files |
| CMS2File | MS2 input file adapter |
| CMSSim | Central class for simulation of mass spectrometry experiments |
| CMapAlignmentAlgorithmIdentification | A map alignment algorithm based on peptide identifications from MS2 spectra |
| CMapAlignmentAlgorithmPoseClustering | A map alignment algorithm based on pose clustering |
| CMapAlignmentAlgorithmSpectrumAlignment | A map alignment algorithm based on spectrum similarity (dynamic programming) |
| CMapAlignmentAlgorithmTreeGuided | A map alignment algorithm based on peptide identifications from MS2 spectra |
| CMascotGenericFile | Read/write Mascot generic files (MGF) |
| CMascotInfile | Mascot input file adapter |
| CMassTraceDetection | A mass trace extraction method that gathers peaks similar in m/z and moving along retention time |
| CMasstraceCorrelator | Correlates individual masstraces found in mass spectrometric maps |
| CMetaboliteSpectralMatching | |
| CMorphologicalFilter | This class implements baseline filtering operations using methods from mathematical morphology |
| CMultiplexClustering | Clusters results from multiplex filtering |
| ►CMultiplexFiltering | Base class for filtering centroided and profile data for peak patterns |
| CMultiplexFilteringCentroided | Filters centroided data for peak patterns |
| CMultiplexFilteringProfile | Filters centroided and profile data for peak patterns |
| CMzDataFile | File adapter for MzData files |
| CMzIdentMLFile | File adapter for MzIdentML files |
| CMzMLFile | File adapter for MzML files |
| CMzQuantMLFile | File adapter for MzQuantML files |
| CMzXMLFile | File adapter for MzXML 3.1 files |
| COpenPepXLAlgorithm | Search for peptide pairs linked with a labeled cross-linker |
| COpenPepXLLFAlgorithm | Search for cross-linked peptide pairs in tandem MS spectra |
| ►COpenSwathWorkflowBase | |
| COpenSwathCalibrationWorkflow | Execute all steps for retention time and m/z calibration of SWATH-MS data |
| ►COpenSwathWorkflow | Execute all steps in an OpenSwath analysis |
| COpenSwathWorkflowSonar | Execute all steps in an OpenEcho analysis (OpenSwath for SONAR data) |
| CPeakPickerCWT | This class implements a peak picking algorithm using wavelet techniques |
| CPeakPickerHiRes | This class implements a fast peak-picking algorithm best suited for high resolution MS data (FT-ICR-MS, Orbitrap). In high resolution data, the signals of ions with similar mass-to-charge ratios (m/z) exhibit little or no overlapping and therefore allow for a clear separation. Furthermore, ion signals tend to show well-defined peak shapes with narrow peak width |
| CPeakPickerIterative | This class implements a peak-picking algorithm for high-resolution MS data (specifically designed for TOF-MS data) |
| CPeakPickerSH | |
| CPeptideIndexing | Refreshes the protein references for all peptide hits in a vector of PeptideIdentifications and adds target/decoy information |
| CQcMLFile | File adapter for QcML files used to load and store QcML files |
| CRawMSSignalSimulation | Simulates MS signals for a given set of peptides |
| CSVMWrapper | Serves as a wrapper for the libsvm |
| CSavitzkyGolayFilter | Computes the Savitzky-Golay filter coefficients using QR decomposition |
| CSignalToNoiseEstimator< Container > | This class represents the abstract base class of a signal to noise estimator |
| CSimpleSearchEngineAlgorithm | |
| CSingleLinkage | SingleLinkage ClusterMethod |
| CSpectraMerger | Merges blocks of MS or MS2 spectra |
| CSwathFile | File adapter for Swath files |
| CTOFCalibration | This class implements an external calibration for TOF data using external calibrant spectra |
| CToolDescriptionFile | File adapter for ToolDescriptor files |
| CTraMLFile | File adapter for HUPO PSI TraML files |
| CTransitionTSVFile | This class supports reading and writing of OpenSWATH transition lists |
| CXFDRAlgorithm | Calculates false discovery rate estimates on crosslink identifications |
| CXMassFile | File adapter for 'XMass Analysis (fid)' files |
| CXQuestResultXMLFile | Used to load and store xQuest result files |
| ►CProgressLogger::ProgressLoggerImpl | This class represents an actual implementation of a logger |
| CGUIProgressLoggerImpl | Implements a GUI version of the ProgressLoggerImpl |
| CProtein | |
| CPeptideAndProteinQuant::ProteinData | Quantitative and associated data for a protein |
| CProteinResolver::ProteinEntry | Protein from FASTA file |
| CIDBoostGraph::ProteinGroup | Placeholder for peptides with the same parent proteins or protein groups |
| CProteinIdentification::ProteinGroup | Bundles multiple (e.g. indistinguishable) proteins in a group |
| CProteinHit::ProteinHitAccessionHash | Hash of a ProteinHit based on its accession only! |
| CProteinHit::ProteinHitPtrAccessionHash | |
| CProteinInference | [experimental class] given a peptide quantitation, infer corresponding protein quantities |
| COPXLDataStructs::ProteinProteinCrossLink | The ProteinProteinCrossLink struct represents a cross-link between two peptides in OpenPepXL |
| CPScore | Implementation of the PScore PSM scoring algorithm |
| CHyperScore::PSMDetail | Compute the (ln transformed) X!Tandem HyperScore overload that returns some additional information on the match |
| CPSProteinInference | This class implements protein inference for the precursor ion selection strategies |
| CIsobaricChannelExtractor::PuritySate_ | Small struct to capture the current state of the purity computation |
| CPrecursorPurity::PurityScores | |
| CPythonInfo | Detect Python and retrieve information |
| ►CQApplication | |
| CQApplicationTOPP | Extension to the QApplication for running TOPPs GUI tools |
| ►CQCBase | This class serves as an abstract base class for all QC classes |
| CContaminants | This class is a metric for the QualityControl TOPP tool |
| CFWHM | QC metric calculating (un)calibrated m/z error |
| CFeatureSummary | |
| CFragmentMassError | |
| CIdentificationSummary | |
| CMissedCleavages | This class is a metric for the QualityControl TOPP Tool |
| CMs2IdentificationRate | This class is a metric for the QualityControl-ToppTool |
| CMs2SpectrumStats | QC metric to determine the number of MS2 scans per MS1 scan over RT |
| CMzCalibration | QC metric calculating (un)calibrated m/z error |
| CPSMExplainedIonCurrent | |
| CPeptideMass | QC metric calculating theoretical mass of a peptide sequence |
| CRTAlignment | Take the original retention time before map alignment and use the alignment's trafoXML for calculation of the new alignment retention times |
| CSpectrumCount | |
| CTIC | |
| ►CQDate | |
| CDate | Date Class |
| ►CQDialog | |
| CDataFilterDialog | Dialog for creating and changing a DataFilter |
| CFeatureEditDialog | Dialog for editing a feature |
| CHistogramDialog | Dialog that show a HistogramWidget |
| CPlot1DPrefDialog | Preferences dialog for Plot1DWidget |
| CPlot2DPrefDialog | Preferences dialog for Plot2DWidget |
| CPlot3DPrefDialog | Preferences dialog for Plot3DWidget |
| CTOPPViewPrefDialog | Preferences dialog for TOPPView |
| CLayerStatisticsDialog | Dialog showing statistics about the data of the current layer |
| CListEditor | Editor for editing int, double and string lists (including output and input file lists) |
| CListFilterDialog | Dialog for creating and changing a DataFilter |
| CMetaDataBrowser | A meta data visualization widget |
| CPlot1DGoToDialog | Simple goto/set visible area dialog for exact placement of the viewing window |
| CPlot2DGoToDialog | GoTo dialog used to zoom to a m/z and retention time range or to a feature |
| CSaveImageDialog | Dialog for saving an image |
| CSpectrumAlignmentDialog | Lets the user select two spectra and set the parameters for the spectrum alignment |
| CTOPPASIOMappingDialog | Dialog which allows to configure the input/output parameter mapping of an edge |
| CTOPPASInputFileDialog | Dialog which allows to specify an input file |
| CTOPPASInputFilesDialog | Dialog which allows to specify a list of input files |
| CTOPPASOutputFilesDialog | Dialog which allows to specify the directory for the output files |
| CTOPPASToolConfigDialog | TOPP tool configuration dialog |
| CTOPPASVertexNameDialog | Dialog which allows to change the name of an input/output vertex |
| CTOPPViewOpenDialog | Dataset opening options for TOPPView |
| CTheoreticalSpectrumGenerationDialog | Dialog which allows to enter an AA sequence and generates a theoretical spectrum for it |
| CToolsDialog | TOPP tool selection dialog |
| ►CQFileSystemWatcher | |
| CFileWatcher | Watcher that monitors file changes |
| ►CQGraphicsItem | |
| CTOPPASEdge | An edge representing a data flow in TOPPAS |
| ►CTOPPASVertex | The base class of the different vertex classes |
| CTOPPASInputFileListVertex | A vertex representing an input file list |
| CTOPPASMergerVertex | A special vertex that allows to merge several inputs |
| CTOPPASOutputFileListVertex | A vertex representing an output file list |
| CTOPPASSplitterVertex | A special vertex that allows to split a list of inputs |
| CTOPPASToolVertex | A vertex representing a TOPP tool |
| ►CQGraphicsScene | |
| CTOPPASScene | A container for all visual items of a TOPPAS workflow |
| ►CQGraphicsView | |
| CTOPPASWidget | Widget visualizing and allowing to edit TOPP pipelines |
| ►CQItemDelegate | |
| CListEditorDelegate | Internal delegate class |
| CParamEditorDelegate | Internal delegate class for QTreeWidget |
| ►CQLineEdit | |
| COpenMSLineEdit | Custom QLineEdit which emits a signal when losing focus (such that we can commit its data) |
| ►CQListWidget | |
| CListTable | |
| CLayerListView | Pimped QListView for Layers of a Canvas |
| ►CQMainWindow | |
| CINIFileEditorWindow | Shows the ParamEditor widget in a QMainWindow with a toolbar |
| CSwathWizardBase | Main window of the SwathWizard tool |
| CTOPPASBase | Main window of the TOPPAS tool |
| CTOPPViewBase | Main window of TOPPView tool |
| ►CQMdiArea | |
| CEnhancedWorkspace | |
| ►CQObject | |
| CExternalProcess | A wrapper around QProcess to conveniently start an external program and forward its outputs |
| CMascotRemoteQuery | Class which handles the communication between OpenMS and the Mascot server |
| CNetworkGetRequest | |
| CRecentFilesMenu | Manages recent files opened by the user and provides a QMenu to go with it |
| CSignalProvider | Signal mechanism (by deriving from QObject) for classes which are not allowed to have signals themselves |
| CTOPPASEdge | An edge representing a data flow in TOPPAS |
| CTOPPASResource | Represents a data resource for TOPPAS workflows |
| CTOPPASResources | A dictionary mapping string keys to lists of TOPPASResource objects |
| CTOPPASVertex | The base class of the different vertex classes |
| CTOPPViewMenu | The file menu items for TOPPView |
| ►CTVControllerBase | Base behavior for different visualizaton modules in TOPPView |
| CTVDIATreeTabController | Behavior of TOPPView in spectra view mode |
| CTVIdentificationViewController | Behavior of TOPPView in identification mode |
| CTVSpectraViewController | Behavior of TOPPView in spectra view mode |
| ►CQOpenGLFunctions_2_0 | |
| CPlot3DOpenGLCanvas | OpenGL Canvas for 3D-visualization of map data |
| ►CQOpenGLWidget | |
| CPlot3DOpenGLCanvas | OpenGL Canvas for 3D-visualization of map data |
| ►CQProcess | |
| CFakeProcess | A FakeProcess class |
| ►CQTabBar | |
| CEnhancedTabBar | Convenience tab bar implementation |
| ►CQTableWidget | |
| CTableView | A better QTable for TOPPView, which supports exporting to TSV and conveniently adding data to cells and headers |
| ►CQTabWidget | |
| CDataSelectionTabs | A tabbed view, to browse lists of spectra or identifications |
| CSwathTabWidget | A multi-tabbed widget for the SwathWizard offering setting of parameters, input-file specification and running Swath and more |
| CQTCluster | A representation of a QT cluster used for feature grouping |
| ►CQTextEdit | |
| CLogWindow | A log window (QTextEdit) with convenience functions |
| ►CQTreeWidget | |
| CParamTree | QTreeWidget that emits a signal whenever a new row is selected |
| CTOPPASTreeView | Tree view implementation for the list of TOPP tools |
| CTreeView | A better QTreeWidget for TOPPView, which supports header context menu and conveniently adding/getting headers names |
| CQuadraticRegression | |
| CBaseFeature::QualityLess | Compare by quality |
| CQcMLFile::QualityParameter | Representation of a quality parameter |
| ►CQWidget | |
| CAxisWidget | Widget that represents an axis of a graph |
| ►CBaseVisualizerGUI | A base class for all visualizer classes |
| CAcquisitionInfoVisualizer | Class that displays all meta information for AcquisitionInfo objects |
| CAcquisitionVisualizer | Class that displays all meta information for Acquisition objects |
| CContactPersonVisualizer | Class that displays all meta information for ContactPerson objects |
| CDataProcessingVisualizer | Class that displays all meta information for DataProcessing objects |
| CDigestionVisualizer | Class that displays all meta information of digestion objects |
| CDocumentIdentifierVisualizer | Class that displays all meta information for DocumentIdentifier objects |
| CExperimentalSettingsVisualizer | Class that displays all meta information for ExperimentalSettings objects |
| CGradientVisualizer | GradientVisualizer is a visualizer class for objects of type gradient |
| CHPLCVisualizer | Class that displays all meta information for HPLC objects |
| CInstrumentSettingsVisualizer | Class that displays all meta information for InstrumentSettings objects |
| CInstrumentVisualizer | Class that displays all meta information for an MS instrument |
| CIonDetectorVisualizer | Class that displays all meta information for IonDetector objects |
| CIonSourceVisualizer | Class that displays all meta information for IonSource objects |
| CMassAnalyzerVisualizer | Class that displays all meta information for MassAnalyzer objects |
| CMetaInfoDescriptionVisualizer | Class that displays all meta information for MetaInfoDescription objects |
| CMetaInfoVisualizer | MetaInfoVisualizer is a visualizer class for all classes that use one MetaInfo object as member |
| CModificationVisualizer | Class that displays all meta information of modification objects |
| CPeptideHitVisualizer | Class that displays all meta information for PeptideHit objects |
| CPeptideIdentificationVisualizer | Class that displays all meta information for PeptideIdentification objects |
| CPrecursorVisualizer | Class that displays all meta information for Precursor objects |
| CProductVisualizer | Class that displays all meta information for Product objects |
| CProteinHitVisualizer | Class that displays all meta information for ProteinHit objects |
| CProteinIdentificationVisualizer | Class that displays all meta information for ProteinIdentification objects |
| CSampleVisualizer | Class that displays all meta information of sample objects |
| CScanWindowVisualizer | Class that displays all meta information for ScanWindow objects |
| CSoftwareVisualizer | Class that displays all meta information for Software objects |
| CSourceFileVisualizer | Class that displays all meta information for SourceFile objects |
| CSpectrumSettingsVisualizer | Class that displays all meta information for SpectrumSettings objects |
| CTaggingVisualizer | Class that displays all meta information of tagging objects |
| CColorSelector | A widget for selecting a color |
| CDIATreeTab | Hierarchical visualization and selection of spectra |
| CHistogramWidget | Widget which can visualize a histogram |
| CInputFile | A simple widget with a line-edit and a browse button to choose filenames |
| CFilterList | A widget which shows a list of DataFilter items |
| CFilterableList | A widget which shows a list of text items, which can be filtered |
| CInputFileList | A widget shows a list of input files (i.e. existing files on a mounted drive), which allows adding/removing files and supports drag'n'drop from the window manager |
| CPythonModuleRequirement | |
| CPythonSelector | |
| CMultiGradientSelector | A widget witch allows constructing gradients of multiple colors |
| COutputDirectory | A simple widget with a line-edit and a browse button to choose filenames |
| CParamEditor | A GUI for editing or viewing a Param object |
| CPlotCanvas | Base class for visualization canvas classes |
| CPlotWidget | Base class for spectrum widgets |
| CSpectraIDViewTab | Tabular visualization / selection of identified spectra |
| CSpectraTreeTab | Hierarchical visualization and selection of spectra |
| CSwathLibraryStats | A multi-tabbed widget for the SwathWizard offering setting of parameters, input-file specification and running Swath and more |
| CRAIICleanup | Exception-safe way of executing arbitrary code at the end of a scope |
| CRandomShuffler | |
| CRange | Internal structure to store a lower and upper bound of an m/z range |
| CRangeManager< D > | Handles the management of a position and intensity range |
| ►CRangeManager< 1 > | |
| CMSChromatogram | The representation of a chromatogram |
| CMSSpectrum | The representation of a 1D spectrum |
| ►CRangeManager< 2 > | |
| CConsensusMap | A container for consensus elements |
| CFeatureMap | A container for features |
| CMSExperiment | In-Memory representation of a mass spectrometry experiment |
| CPeptideHit::RankLess | Lesser predicate for scores of hits |
| CRANSAC< TModelType > | This class provides a generic implementation of the RANSAC outlier detection algorithm. Is implemented and tested after the SciPy reference: http://wiki.scipy.org/Cookbook/RANSAC |
| CRansacModel< ModelT > | Generic plug-in template base class using 'Curiously recurring template pattern' (CRTP) to allow for arbitrary RANSAC models (e.g. linear or quadratic fits) |
| ►CRansacModel< RansacModelLinear > | |
| CRansacModelLinear | Implementation of a linear RANSAC model fit |
| ►CRansacModel< RansacModelQuadratic > | |
| CRansacModelQuadratic | Implementation of a quadratic RANSAC model fit |
| CRANSACParam | A simple struct to carry all the parameters required for a RANSAC run |
| CConsensusFeature::Ratio | Slim struct to feed the need for systematically storing of ratios ( |
| ►Creal_policies | |
| CBK_PrecPolicy< T > | |
| ►Creal_policies | |
| CStringUtils::real_policies_NANfixed_< T > | |
| CRealMassDecomposer | Handles decomposing of non-integer values/masses over a set of non-integer weights with an error allowed |
| CBase64::Reinterpreter32_ | Internal class needed for type-punning |
| CBase64::Reinterpreter64_ | Internal class needed for type-punning |
| CResidue | Representation of a residue |
| CResidueDB | OpenMS stores a central database of all residues in the ResidueDB. All (unmodified) residues are added to the database on construction. Modified residues get created and added if getModifiedResidue is called |
| CResidueModification | Representation of a modification |
| CProteinResolver::ResolverResult | |
| CDecoyHelper::Result | |
| CFeatureSummary::Result | |
| CIdentificationSummary::Result | |
| CMorpheusScore::Result | Score and subscores |
| CTIC::Result | |
| CReverseComparator< Cmp > | Wrapper that reverses (exchanges) the two arguments of a comparator. Normally you should use the make-function reverseComparator() because then you do not need to specify the template arguments |
| CRibonucleotide | Representation of a ribonucleotide (modified or unmodified) |
| CRibonucleotideDB | Database of ribonucleotides (modified and unmodified) |
| CRNPxlFragmentAnnotationHelper | Convenience functions to construct appealing fragment annotation strings and store them as PeptideHit::PeakAnnotation |
| CRNPxlMarkerIonExtractor | |
| CRNPxlModificationMassesResult | |
| CRNPxlModificationsGenerator | |
| CRNPxlReport | Create report |
| CRNPxlReportRow | Struct to hold a single report line |
| CRNPxlReportRowHeader | Create header line |
| CROCCurve | ROCCurves show the trade-off in sensitivity and specificity for binary classifiers using different cutoff values |
| CMzTabNucleicAcidSectionRow::RowCompare | Comparison operator for sorting rows |
| CMzTabOligonucleotideSectionRow::RowCompare | Comparison operator for sorting rows |
| CMzTabOSMSectionRow::RowCompare | Comparison operator for sorting rows |
| CMzTabPeptideSectionRow::RowCompare | Comparison operator for sorting rows |
| CMzTabProteinSectionRow::RowCompare | Comparison operator for sorting rows |
| CMzTabPSMSectionRow::RowCompare | Comparison operator for sorting rows |
| CChromatogramPeak::RTLess | Comparator by RT position |
| CMSSpectrum::RTLess | Comparator for the retention time |
| CMZTrafoModel::RTLess | Comparator by position. As this class has dimension 1, this is basically an alias for MZLess |
| CPeak2D::RTLess | Comparator by RT position |
| COSSpectrumMeta::RTLess | Comparator for the retention time |
| CConfidenceScoring::RTNorm_ | Helper for RT normalization (range 0-100) |
| CFeatureFinderIdentificationAlgorithm::RTRegion | Region in RT in which a peptide elutes: |
| CAbsoluteQuantitationStandards::runConcentration | Structure to map runs to components to known concentrations |
| CRWrapper | R-Wrapper Class |
| CExperimentalDesign::SampleSection | |
| CMs2SpectrumStats::ScanEvent | |
| CPSLPFormulation::ScanLess | |
| CPeptideHit::ScoreLess | Lesser predicate for scores of hits |
| CProteinHit::ScoreLess | Lesser predicate for scores of hits |
| CPeptideHit::ScoreMore | Greater predicate for scores of hits |
| CProteinHit::ScoreMore | Greater predicate for scores of hits |
| CFeatureFinderAlgorithmPickedHelperStructs::Seed | Helper structure for seeds used in FeatureFinderAlgorithmPicked |
| CSeedListGenerator | Generate seed lists for feature detection |
| CMRMFeatureSelector::SelectorParameters | |
| CPrecursorIonSelection::SeqTotalScoreMore | Compare by score |
| CPeptideHit::SequenceLessComparator | Lesser predicate for (modified) sequence of hits |
| CSequestInfile | Sequest input file adapter |
| CSequestOutfile | Representation of a Sequest output file |
| ►Cset< K > | STL class |
| CIsotopeCluster::ChargedIndexSet | Index set with associated charge estimate |
| Cshared_xerces_ptr< T > | |
| CSignalToNoiseEstimatorMedianRapid | Estimates the signal/noise (S/N) ratio of each data point in a scan by using the median (window based) |
| CSimpleOpenMSSpectraFactory | A factory method that returns two ISpectrumAccess implementations |
| CSimpleTSGXLMS::SimplePeak | A simple struct to represent peaks with mz and charge and sort them easily |
| CSimpleTSGXLMS::SimplePeakComparator | Comparator to sort SimplePeaks by mz |
| CSimProtein | Plain data object holding sequence and abundance information on a single protein |
| CSimRandomNumberGenerator | Wrapper class for random number generators used by the simulation classes |
| CROCCurve::simsortdec | Predicate for sort() |
| CSingletonRegistry | Holds pointers to unique instance of a singleton factory |
| CSiriusMzTabWriter::SiriusAdapterHit | Internal structure used in SiriusAdapter that is used for the conversion of the sirius output to an mzTab |
| CSiriusMzTabWriter::SiriusAdapterIdentification | |
| CSiriusMzTabWriter::SiriusAdapterRun | |
| CSiriusFragmentAnnotation | |
| CSiriusMSFile | |
| CSiriusMzTabWriter | |
| CSiriusMzTabWriter::SiriusSpectrumMSInfo | |
| CSiriusFragmentAnnotation::SiriusTargetDecoySpectra | SiriusTargetDecoySpectra holds the target and/or decoy information for one entry (subdirectory from SIRIUS) |
| CSiriusAdapterAlgorithm::SiriusTemporaryFileSystemObjects | Struct for temporary folder structure |
| CConsensusFeature::SizeLess | Compare by size(), the number of consensus elements |
| CLPWrapper::SolverParam | Struct that holds the parameters of the LP solver |
| CSpawn< TNeedle > | State of an AC spawn, operating on a trie |
| CSpecArrayFile | File adapter for SpecArray (.pepList) files |
| CMzMLFile::SpecInfo | |
| CIDMapper::SpectraIdentificationState | Result of a partitioning by identification state with mapPrecursorsToIdentifications() |
| CSpectralMatch | |
| CSpectralMatchScoreComparator | |
| CQCBase::SpectraMap | Map to find a spectrum via its NativeID |
| CSpectrum | The structure that captures the generation of a peak list (including the underlying acquisitions) |
| CSpectrumAddition | The SpectrumAddition is used to add up individual spectra |
| CMzMLHandler::SpectrumData | Data necessary to generate a single spectrum |
| CMzXMLHandler::SpectrumData | Data necessary to generate a single spectrum |
| CMzIdentMLDOMHandler::SpectrumIdentification | Struct to hold the information from the SpectrumIdentification xml tag |
| CMzIdentMLDOMHandler::SpectrumIdentificationProtocol | Struct to hold the information from the SpectrumIdentificationProtocol xml tag |
| ►CSpectrumLookup | Helper class for looking up spectra based on different attributes |
| CSpectrumMetaDataLookup | Helper class for looking up spectrum meta data |
| CSpectrumMeta | Identifying information for a spectrum |
| CSpectrumMetaDataLookup::SpectrumMetaData | Meta data of a spectrum |
| CSplineInterpolatedPeaks | Data structure for spline interpolation of MS1 spectra and chromatograms |
| CSplinePackage | Fundamental data structure for SplineInterpolatedPeaks |
| CSqliteConnector | File adapter for Sqlite files |
| CSqMassFile::SqMassConfig | Configuration class for SqMassFile |
| CSqMassFile | An class that uses on-disk SQLite database to read and write spectra and chromatograms |
| Csquared_difference< _Tp, _Dist > | |
| Csquared_difference< typename _Acc::result_type, typename _Acc::result_type > | |
| Csquared_difference_counted< _Tp, _Dist > | |
| ►Cstatic_visitor | |
| CIDBoostGraph::GetPosteriorVisitor | |
| CIDBoostGraph::LabelVisitor | Visits nodes in the boost graph (ptrs to an ID Object) and depending on their type creates a label |
| CIDBoostGraph::PrintAddressVisitor< CharT > | |
| CIDBoostGraph::SetPosteriorVisitor | |
| CFragmentMassError::Statistics | Structure for storing results: average and variance of all FragmentMassErrors in ppm |
| CPeptideAndProteinQuant::Statistics | Statistics for processing summary |
| CPSMExplainedIonCurrent::Statistics | Structure for storing results: average and variance over all PSMs |
| CStopWatch | This class is used to determine the current process' CPU (user and/or kernel) and wall time |
| ►Cstreambuf | |
| CLogStreamBuf | Stream buffer used by LogStream |
| CFuzzyStringComparator::StreamElement_ | Stores information about characters, numbers, and white spaces loaded from the InputStream |
| CStreamHandler | Provides a central class to register globally used output streams. Currently supported streams are |
| CLogStreamBuf::StreamStruct | Holds a stream that is connected to the LogStream. It also includes the minimum and maximum level at which the LogStream redirects messages to this stream |
| CString< AAcid, Alloc< void > > | |
| CStringListUtils | Utilities operating on lists of Strings |
| CStringManager | |
| CStringUtils | |
| CStringView | StringView provides a non-owning view on an existing string |
| CDBSuitability::SuitabilityData | Struct to store results |
| CSummary | Summary of fitting results |
| CSummaryStatistics< T > | Helper class to gather (and dump) some statistics from a e.g. vector<double> |
| CTargetedExperiment::SummaryStatistics | |
| CSVMData | Data structure used in SVMWrapper |
| CSvmTheoreticalSpectrumGenerator::SvmModelParameterSet | Simple container storing the model parameters required for simulation |
| CSvmTheoreticalSpectrumGeneratorSet | Loads SvmTheoreticalSpectrumGenerator instances for different charges |
| CSwathGUILock | RAII class to switch to certain TabWidget, disable the GUI and go back to the orignal Tab when this class is destroyed |
| CSwathMap | Data structure to hold one SWATH map with information about upper / lower isolation window and whether the map is MS1 or MS2 |
| CSwathQC | Quality Control function for OpenSwath |
| CSwathWindowLoader | Class to read a file describing the Swath Windows |
| CSysInfo | Some functions to get system information |
| CTagger | Calculate sequence tags from m/z values |
| CMetaboTargetedAssay::TargetDecoyGroup | TargetDecoyGroup stores the mz, rt and file number in correspondence to the index of a MetaboTargetedAssay vector |
| CTargetedExperiment | A description of a targeted experiment containing precursor and production ions |
| CTargetedExperiment | |
| CFile::TempDir | Class representing a temporary directory |
| CFile::TemporaryFiles_ | Internal helper class, which holds temporary filenames and deletes these files at program exit |
| ►CTextFile | This class provides some basic file handling methods for text files |
| ►CCsvFile | This class handles csv files. Currently only loading is implemented |
| CAbsoluteQuantitationMethodFile | File adapter for AbsoluteQuantitationMethod files |
| CMRMFeaturePickerFile | _MRMFeaturePickerFile_ loads components and components groups parameters from a .csv file |
| CMRMFeatureQCFile | File adapter for MRMFeatureQC files |
| CFeatureFinderAlgorithmPickedHelperStructs::TheoreticalIsotopePattern | Helper structure for a theoretical isotope pattern used in FeatureFinderAlgorithmPicked |
| CStopWatch::TimeDiff_ | |
| ►CToolDescriptionInternal | ToolDescription Class |
| CToolDescription | |
| CToolExternalDetails | |
| CToolHandler | |
| CTOPPASVertex::TOPPASFilenames | |
| ►CTOPPBase | Base class for TOPP applications |
| CNucleicAcidSearchEngine | |
| CSearchEngineBase | Base class for Search Engine Adapters |
| CTOPPOpenSwathBase | |
| CTOPPGNPSExport | |
| CTOPPASScene::TOPPProcess | Stores the information for a TOPP process |
| CPrecursorIonSelection::TotalScoreMore | Compare by score |
| CParam::ParamIterator::TraceInfo | Struct that captures information on entered / left nodes for ParamIterator |
| CIDDecoyProbability::Transformation_ | Struct to be used to store a transformation (used for fitting) |
| CTransformationDescription | Generic description of a coordinate transformation |
| ►CTransformationModel | Base class for transformation models |
| CTransformationModelBSpline | B-spline (non-linear) model for transformations |
| CTransformationModelInterpolated | Interpolation model for transformations |
| CTransformationModelLinear | Linear model for transformations |
| CTransformationModelLowess | Lowess (non-linear) model for transformations |
| CTransformationDescription::TransformationStatistics | Summary statistics before/after applying the transformation. For deviations before/after transformation, the percentiles 100, 99, 95, 90, 75, 50, 25 are returned |
| CTransitionHelper | |
| CIsotopeWaveletTransform< PeakType >::TransSpectrum | Internally (only by GPUs) used data structure . It allows efficient data exchange between CPU and GPU and avoids unnecessary memory moves. The class is tailored on the isotope wavelet transform and is in general not applicable on similar - but different - situations |
| CTriqlerFile | File adapter for Triqler files |
| CTriqlerFile::TriqlerLine_ | |
| CTransitionTSVFile::TSVTransition | Internal structure to represent a transition |
| CTVToolDiscovery | Scans for tools/utils and generates a param for each asynchronously |
| CTwoDOptimization::TwoDOptFunctor | |
| Cunique_xerces_ptr< T > | |
| CIdentificationSummary::UniqueID | |
| CUniqueIdGenerator | A generator for unique ids |
| CUniqueIdIndexer< RandomAccessContainer > | A base class for random access containers for classes derived from UniqueIdInterface that adds functionality to convert a unique id into an index into the container |
| ►CUniqueIdIndexer< ConsensusMap > | |
| CConsensusMap | A container for consensus elements |
| ►CUniqueIdIndexer< FeatureMap > | |
| CFeatureMap | A container for features |
| ►CUniqueIdInterface | A base class defining a common interface for all classes having a unique id |
| CConsensusMap | A container for consensus elements |
| CFeatureHandle | Representation of a Peak2D, RichPeak2D or Feature |
| CFeatureMap | A container for features |
| CRichPeak2D | A 2-dimensional raw data point or peak with meta information |
| CCVTerm::Unit | |
| CUpdateCheck | Helper Functions to perform an update query to the OpenMS REST server |
| CValueSize< AAcid > | |
| CValueTrait | Trait for MatchedIterator to find pairs with a certain distance, which is computed directly on the value_type of the container |
| CPSLPFormulation::VariableIndexLess | |
| CVecLowPrecision< T > | |
| ►Cvector< T > | STL class |
| CMatrix< double > | |
| CMatrix< UInt > | |
| CAcquisitionInfo | Description of the combination of raw data to a single spectrum |
| CConsensusMap | A container for consensus elements |
| CFloatDataArray | Float data array class |
| CIntegerDataArray | Integer data array class |
| CStringDataArray | String data array class |
| CFeatureFinderAlgorithmPickedHelperStructs::MassTraces | Helper struct for a collection of mass traces used in FeatureFinderAlgorithmPicked |
| CFeatureMap | A container for features |
| CMSChromatogram | The representation of a chromatogram |
| CMSSpectrum | The representation of a 1D spectrum |
| CMatrix< Value > | A two-dimensional matrix. Similar to std::vector, but uses a binary operator(,) for element access |
| CScoreToTgtDecLabelPairs | |
| CVersionInfo::VersionDetails | |
| CVersionInfo | Version information class |
| CTOPPASVertex::VertexRoundPackage | Info for one edge and round, to be passed to next node |
| CWeights | Represents a set of weights (double values and scaled with a certain precision their integer counterparts) with a quick access |
| CWeightWrapper | Encapsulated weight queries to simplify mono vs average weight computation |
| CInclusionExclusionList::WindowDistance_ | Determine distance between two spectra |
| CXCorrArrayType | |
| COPXLDataStructs::XLPrecursor | The XLPrecursor struct represents a cross-link candidate in the process of filtering candidates by precursor masses in OpenPepXL |
| COPXLDataStructs::XLPrecursorComparator | The XLPrecursorComparator is a comparator for XLPrecursors, that allows direct comparison of the XLPrecursor precursor mass with double numbers |
| ►CXMLFile | Base class for loading/storing XML files that have a handler derived from XMLHandler |
| CCVMappingFile | Used to load CvMapping files |
| CConsensusXMLFile | This class provides Input functionality for ConsensusMaps and Output functionality for alignments and quantitation |
| CFeatureXMLFile | This class provides Input/Output functionality for feature maps |
| CIdXMLFile | Used to load and store idXML files |
| CSemanticValidator | Semantically validates XML files using CVMappings and a ControlledVocabulary |
| CMascotXMLFile | Used to load Mascot XML files |
| CMzDataFile | File adapter for MzData files |
| CMzIdentMLFile | File adapter for MzIdentML files |
| CMzMLFile | File adapter for MzML files |
| CMzQuantMLFile | File adapter for MzQuantML files |
| CMzXMLFile | File adapter for MzXML 3.1 files |
| COMSSAXMLFile | Used to load OMSSAXML files |
| CPTMXMLFile | Used to load and store PTMXML files |
| CParamXMLFile | The file pendant of the Param class used to load and store the param datastructure as paramXML |
| CPepXMLFile | Used to load and store PepXML files |
| CPepXMLFileMascot | Used to load Mascot PepXML files |
| CProtXMLFile | Used to load (storing not supported, yet) ProtXML files |
| CQcMLFile | File adapter for QcML files used to load and store QcML files |
| CToolDescriptionFile | File adapter for ToolDescriptor files |
| CTraMLFile | File adapter for HUPO PSI TraML files |
| CTransformationXMLFile | Used to load and store TransformationXML files |
| CUnimodXMLFile | Used to load XML files from unimod.org files |
| CXQuestResultXMLFile | Used to load and store xQuest result files |
| CXTandemInfile | XTandem input file |
| CXTandemXMLFile | Used to load XTandemXML files |
| CXQuestScores | An implementation of the scores for cross-link identification from the xQuest algorithm (O. Rinner et al., 2008, "Identification of cross-linked peptides from large sequence databases") |
| CZlibCompression | Compresses and uncompresses data using zlib |
 1.9.1
1.9.1

